In the last decade, cancer became the leading cause of death in the population under 65 in the European Union. Diabetes is also considered as a factor increasing risk of cancer incidence and mortality. Type 2 diabetes is frequently associated with being overweight and obese, which also plays a role in malignancy. Among biological mechanisms linking diabetes and obesity with cancer hyperglycemia, hyperinsulinemia, insulin resistance, increased levels of growth factors, steroid and peptide hormones, oxidative stress and increased activity of pro-inflammatory cytokines are listed. Antidiabetic medications can modulate cancer risk through directly impacting metabolism of cancer cells as well as indirectly through impact on risk factors of malignancy. Some of them are considered beneficial (metformin and thiazolidinedions—with the exception of bladder cancer); on the other hand, excess of exogenous insulin may be potentially harmful, while other medications seem to have neutral impact on cancer risk. Inhibitors of the sodium-glucose cotransporter-2 (SGLT-2) are increasingly used in the treatment of type 2 diabetes. However, their association with cancer risk is unclear.
- Antidiabetic Medications
- Cancer
- Metformin
- Sulfonylureas
- Thiazolidinedions
- Insulin
- DPP-4 inhibitors
- GLP-1 receptor agonists
- SGLT-2 inhibitors
Due to its progressive nature, type 2 diabetes requires a gradual intensification of treatment, starting usually with monotherapy, through the use of 2–3 oral medications, up to injectable therapies with the use of GLP-1 (Glucagon-like peptide-1) receptor agonists and/or various insulin therapy regimens, with or without oral medications [1][2]. Therefore, it is not easy to determine the individual effect of particular antidiabetic drugs on the risk of cancer, due to the long time of cancer development and the changing therapeutic models over time [3]. Moreover, an interplay between antidiabetic medications, can also have an influence on cancer risk. Nevertheless, many data, mainly from observational studies (randomized clinical trials are usually too short to draw firm conclusions on the risk of cancer), indicate the protective effect of metformin and the pro-oncogenic effect of exogenous insulin. According to current data, other classes of drugs may carry a risk of site-specific cancers, but longer follow-up is necessary to confirm or rule out such relationships.
1. Metformin
The first clinical reports indicating the protective effect of metformin in terms of cancer incidence and cancer death risk appeared in the middle of the first decade of this century [4][5]. Own studies also confirmed protective effect of metformin in the Polish population, irrespective of gender [6][7]. In numerous meta-analyses of case-control and cohort studies, metformin use was associated with 10–40% reduction of cancer incidence and mortality, which was summarized by Heckman-Stoddard et al. in their paper [8]. Schulten in his review documented beneficial effect of metformin in many site-specific cancers. Metformin was associated with lower incidence of gastric, colorectal, liver, breast and endometrial cancers, and with longer survival/lower mortality from colorectal, lung, breast, endometrial, prostate and pancreatic cancers [9].
One of the mechanisms of the antihyperglycemic action of metformin is inhibition of the activity of complex I of the mitochondrial respiratory chain, which reduces the availability of ATP and inhibits gluconeogenesis. This process requires the activation of AMP-activated protein kinase (AMPK). Phosphorylation of AMPK occurs in the presence of LKB1 (Liver kinase B1), a tumor suppressor gene product, and is facilitated by AMP. Activation of AMPK leads to the inhibition of the mTOR (mechanistic target of rapamycin) signaling protein. The second site of antihyperglycemic action of metformin is gut, where metformin increases both glucose utilization as well as lactate production by enterocytes, which contributes considerably in maintaining glucose homeostasis [10].
However, inhibition of mTOR leads not only to inhibition of gluconeogenesis in hepatocytes, but also reduces protein synthesis and inhibits the proliferation of neoplastic cells, inducing their apoptosis and cycle-cell arrest [11][12]. Anti-tumor effect of metformin was also documented by Sanaki et al. [13]. AMPK-dependent pathway is considered to play important role in anticancer effect of metformin in several site-specific cancers, e.g., breast [14], endometrial [15] or colorectal cancers [16]. Moreover, this mechanism also plays a role in mitigating DPP-4 inhibitor-induced breast cancer metastasis [17].
Metformin can also inhibit cancer growth indirectly. Decreased hepatic glucose production and increased peripheral glucose uptake, mainly by muscle cells, lead to reduced insulin release from pancreatic β-cells and decreased plasma insulin level, thus reducing risk of proliferation of neoplastic and pre-neoplastic cells [11]. Schulten in his review also discusses several other pathways leading to decreased cancer risk associated with metformin. Metformin inhibits transforming growth factor beta 1 (TGF-β1) thus leading to inhibition of epithelial-to-mesenchymal transition (EMT); it also inhibits cyclin D1 leading to cell cycle arrest; it inhibits AKT serine/threonine kinase 1 leading to growth inhibition; it promotes immune response through protection of CD8+ tumor infiltrating lymphocytes and accumulation of M1-like macrophages which reduces cancer growth and angiogenesis; it inhibits signal transducer and activator of transcription 3 (p-STAT 3) thus activating apoptotic and autophagous pathway through downregulating BLC 2, apoptosis regulator; it also upregulates some micro RNAs which inhibits cancer cells growth and impairs cancer cells viability; it inhibits vascular endothelial growth factor (VEGF) thus inhibiting angiogenesis; it also leads to cancer stem cells depletion through different signaling pathways. Metformin can also inhibit mTOR pathway through DNA damage inducible transcript 4 (DDIT 4) thus inhibiting protein synthesis in cancer cell. Moreover, metformin combined with other chemotherapeutic drugs demonstrates additive or synergistic effect to enhance its anticancer action [9].
2. Sulfonylurea Derivatives
Drugs of this class, by binding to a specific sulfonylurea receptor 1 (SUR1) on the β-cell surface, cause exocytosis of insulin accumulated in secretory granules [18]. This leads to an increase in endogenous insulin levels and may theoretically be associated with an increased risk of cancer. The first observational studies seemed to confirm this relationship [4][5][19], although there were differences between individual formulations [20]. Own observation, as well as the largest systematic review, did not show an increased risk of cancer associated with the use vs. non-use of sulfonylureas [6][7][21].
3. Insulin
The first observation of an association between the use of exogenous insulin and the risk of colorectal cancer dates back to 2004 [22]. Subsequent studies have confirmed a higher incidence of cancer and a higher risk of death from cancer in people using insulin [4][5][19]. The controversial (due to the methodology used) work by Hemkens et al. showed a higher risk of cancer in patients using long-acting insulin analog glargine compared to human insulin. However, it also indicated a much more significant dose-dependent increase in the risk of cancer regardless of the type of insulin used [23]. In the following years, many studies analyzing the relationship between exogenous insulin and cancer were published. A meta-analysis performed by Karlstad et al. revealed a significant 52% increase in cancer risk in people treated with insulin [24]. Insulin monotherapy was associated with elevated risk of cancer incidence in a dose-dependent manner in a study by Holden et al. [25]. Own study also showed elevated risk of cancer associated with insulin monotherapy, but it was attenuated to insignificant level when insulin was combined with metformin [26]. On the other hand, in prospective, randomized clinical trial (RCT), Outcome Reduction with an Initial Glargine Intervention (ORIGIN) trial, elevated cancer risk among insulin users has not been found [27]. However, in this study relatively low doses of insulin were used, while in a real-life setting, insulin therapy is usually initiated in patients with longer-lasting diabetes, worse metabolic control and in older age compared to patients treated with other therapeutic regimens. Frequently, large doses of insulin are used to attain and maintain glycemic control. Such a cluster of risk factors of malignancy can significantly influence cancer risk. Thus, due to a mitogenic effect of exogenous insulin in a mechanism similar to endogenous insulin, its overdosing, generating iatrogenic hyperinsulinemia, should be avoided.
4. Thiazolidinedions
Agonists of the γ isoform of the peroxisome proliferator-activated receptor gamma (PPAR-γ), a nuclear hormone receptor, are a group of antidiabetic drugs that have been present on the market for a several years. Their main site of action is adipose tissue, where they improve insulin sensitivity and inhibit the production of pro-inflammatory cytokines [28]. Their multiple side effects (weight gain, increased risk of heart failure, edema and bone fractures) limit their use in therapy. In the meta-analysis of 119 studies use of glitazones was associated with significant reduction of overall cancer risk [29]. Glitazones exerts its anticancer effect in several mechanisms: cell growth arrest through decreasing activity of S-phase kinase-associated protein 2 (Skp2), a ubiquitin ligase, which leads to upregulation of p27kip1, a cyclin-dependent kinase inhibitor; induction of apoptosis through upregulation of genes p53, PTEN and BAX (BCL2 Associated X), apoptosis regulator, and downregulation of bcl-2/bcl-xL and survivin, antiapoptotic molecules; and inhibition of invasion by inhibition of MEK/ERK signaling pathway which leads to increased expression of E-cadherin and claudin-4 [30]. However, in people using pioglitazone, a higher risk of bladder cancer was found in meta-analysis of RCTs and observational studies. In case of observational studies this association was time- and dose-dependent. It is hypothesized by the authors, that this phenomenon is associated with high expression of PPAR-γ in bladder cells in which their activation is associated with tumor growth and progression [31]. A lower dose of pioglitazone seems to have less adverse effects, while not compromising its beneficial effects on blood glucose, insulin sensitivity and cancer [30].
5. Dipeptidil-Peptidase-4 Inhibitors
Dipeptidil-peptidase-4 (DPP-4) is an enzyme involved in degradation of many substrates including growth factors, chemokines, neuropeptides, vasoactive peptides and incretin hormones-glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). GLP-1 and GIP by binding to specific receptors on the surface of pancreatic β-cells, stimulate insulin release in response to meal, mainly reach in carbohydrate content. Thus, inhibition of their degradation by DPP-4 inhibitors increases incretin hormones activity and has an impact on glucose metabolism which is used in type 2 diabetes treatment [32]. The animal studies suggested that inhibition of DPP-4 may be associated with elevated risk of pancreatic, ovarian, prostate, skin, and lung cancers [33]. Moreover, it was also postulated that DPP-4 inhibition can increase the risk of metastasis through induction of CXCL12/CXCR4, which activates mTOR to promote epithelial–mesenchymal transition (EMT) [34]. On the basis of the US Food and Drug Administration’s database of adverse event reporting system (AERS), based on voluntary reports sent by physicians, Elashoff M et al. in 2011, revealed increased risk of pancreatitis and pancreatic cancer associated with the use of sitagliptin, a DPP-4 inhibitor [35]. These data raised concerns about DPP-4 inhibitors long-term safety. However, later years did not confirm these findings. Meta-analysis of site-specific cancers associated with DPP-4 inhibitors use in 12 RCTs and 13 observational studies did not reveal elevated cancer risk in DPP-4 inhibitors’ users [36]. The most recent meta-analysis of 157 RCTs, performed by Dicembrini and colleagues, revealed neutral effect of DPP-4 inhibitors on overall cancer risk, irrespective of molecule studied and cancer site, with the exception of colorectal cancer, in which DPP-4 inhibitors use was associated with its significantly reduced risk [37].
6. Glucagon-Like Peptide-1 Receptor Agonists
Similar to sitagliptin, Elashoff M et al. revealed elevated risk of pancreatitis, as well as pancreatic and thyroid cancers in patients treated with exenatide, a GLP-1 receptor agonist (GLP-1 RA) [35]. These findings also raised concerns regarding safety of this class of drugs. In fact, stimulation of the GIP and GLP-1 receptor, apart from insulinotropic effects, also activates anti-apoptotic and pro-proliferative processes. The final effect of their stimulation is the activation of the pathway leading to the activation of Mek 1/2 (MAPK/ERK kinase 1/2) and ERK 1/2 (extracellular signal-regulated kinase), which, after entering to the nucleus, catalyzes the phosphorylation of transcription factors leading to cell proliferation and differentiation. It also inhibits the activity of caspase-3, an enzyme involved in the apoptotic process. Activation of GIP and GLP-1 receptors stimulates the proliferation of not only β-cells but also their progenitor cells. In islet cell culture, both GIP and GLP-1 induced the transcription of the cyclin D1 gene, an enzyme that is crucial for the initiation of mitosis in most cell types. In addition, activation of the canonical β-catenin/Wnt signaling pathway was observed [38][39][40]. Moreover, GLP-1 receptors are also present in many other tissues, including thyroid, exocrine pancreas, liver, hypothalamus, renal tubules, and bone, and their activation produces effects completely unrelated to glucose homeostasis [35].
Further observations did not confirm elevated risk of cancer in GLP-1 RA users. Cao C et al. in the most recent meta-analysis of the 37 RCTs did not reveal elevated risk of pancreatic, thyroid and overall cancer in GLP-1 RA users compared to controls [41].
7. Sodium-Glucose Cotransporter-2 Inhibitors
Different types of sodium-glucose cotransporters are present in the entire body. Sodium-glucose cotransporter-2 (SGLT-2) is mainly present in the S1 segment of the proximal convoluted tubule in the kidney (but it was also found in the pancreas, brain, liver, thyroid, muscle and heart). Its main role is reabsorption of glucose from glomerular filtrate [42]. SGLT-1 is also present in the kidney (in the S3 segment of the proximal convoluted tubule), but the main location of its action is the small intestine [42][43]. SGLT-2 and SGLT-1 play important role in maintaining glucose homeostasis. Inhibition of SGLT-2 leads to increased glucose excretion with urine and offers glucose lowering effect independent of insulin [43]. SGLT-2 inhibitors in the CardioVascular Outcome Trials (CVOTs) and renal outcome trials showed, in addition to glucose-lowering effect, body weight and blood pressure reduction, a wide pleiotropic effect, resulting in a reduction of the risk of cardiovascular events, heart failure and kidney disease progression [44][45][46][47][48][49][50][51][52][53]. This resulted in far-reaching changes in scientific associations’ clinical practice recommendations and enabled a wide entrance of these compounds to the market of antidiabetic medications [1]. However, at the beginning of SGLT-2 inhibitors use, concerns were raised about an increased risk of bladder and breast cancers, which led to the rejection of an application for approval of dapagliflozin by Food and Drug Administration in 2011 [54]. In addition, early meta-analysis performed by Vasilakou et al. revealed imbalanced incidence of bladder and breast cancer in dapagliflozin users compared to controls [55]. More recent meta-analysis of 46 RCTs performed by Tang et al. did not reveal overall increased cancer risk. However, it maintained concerns about an increased risk of bladder cancer. On the other hand, the total number of cases (18/22,359 vs. 1/12,228) was too low to draw far-reaching conclusions. Interestingly, canagliflozin use was associated with lower risk of gastrointestinal cancers, which was not observed for dapagliflozin and empagliflozin [56]. The most recent meta-analysis of 27 RCTs performed by Dicembrini et al. did not reveal any difference in cancer incidence between SGLT-2 inhibitors and comparators, including placebo [57].
Based on the data from published cardiovascular and renal outcome trials [44][45][46][47][48][49][50][52][53][58], I performed a meta-analysis of the risk of neoplasms incidence in those trials. I requested the data regarding incidence of malignancies in the Empagliflozin Outcome Trial in Patients with Chronic Heart Failure and a Reduced Ejection Fraction (EMPEROR Reduced), but I did not receive these data and it was not included into meta-analysis. Statistical analysis was performed using PQStat v.1.8.0 software (PQStat Software, Poznań, Poland). A random effects model (Mantel-Haenszel method) was used to calculate pooled Risk Ratios (RR). Overall risk of neoplasm incidence appeared to be insignificant, HM-RR 1.11 (95% confidence interval 0.98–1.26), p = 0.102. The RRs for each study and their summary are presented as the forest plot (Figure 1).
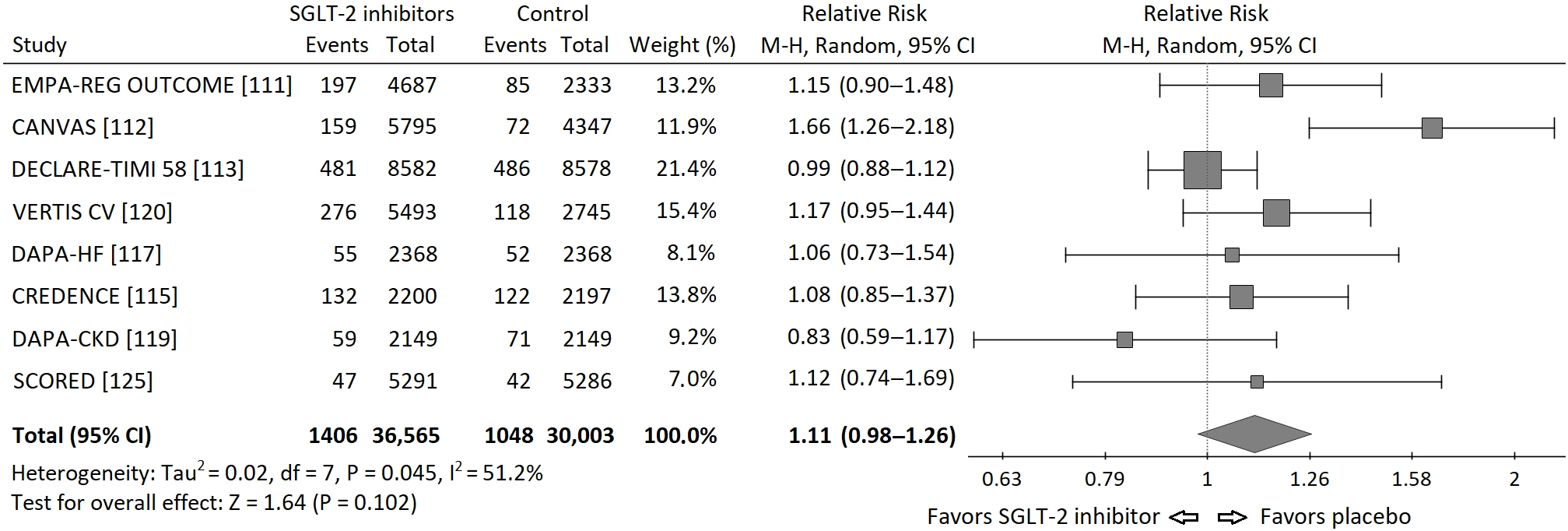
Figure 1. Forest plot of meta-analysis of the risk of neoplasms incidence for SGLT-2 inhibitors vs. placebo in cardiovascular and renal outcome trials. The size of the square box is proportional to the weight that each study contributes in the meta-analysis. The overall estimate and CI are marked by a diamond.
A moderate but significant heterogeneity between the studies was found, I2 = 51.2%, Q-statistics 14.35, p value 0.045. It is worth to note, that there were important differences between populations included in particular studies. In the Dapagliflozin in Patients With Heart Failure and Reduced Ejection Fraction (DAPA-HF) trial exclusively patients with heart failure and reduced ejection fraction (EF) below 40% were included [49], while in the renal outcome trials: Canagliflozin and Renal Events in Diabetes With Established Nephropathy Clinical Evaluation (CREDENCE) and Dapagliflozin and Prevention of Adverse-outcomes in CKD (DAPA-CKD), patients had chronic kidney disease (CKD) mainly at stages 3 and 4 of (⅔ and 90% respectively) [47][51]. Moreover, the duration of each study was different (from 1.2 to 4.2 years). In addition, in DAPA-HF and DAPA-CKD trials also patients without diabetes were included. Thus, the risk of neoplasm incidence was different in particular studies, which can, at least partly, explain the significant heterogeneity found in the analysis.
There are several mechanisms linking SGLT-2 inhibitors with cancer risk. Glucose is the main source of energy for cancer cells. It enters into the cell by using glucose transporters, mainly GLUT 1, which are frequently overexpressed in cancer cells. However, also presence or overexpression of sodium-glucose cotransporters have been identified on the surface of several site-specific cancer cells. Both SGLT-1 and SGLT-2 are present in pancreatic, brain and prostate cancers, in addition SGLT-1 is present in ovary, head and neck cancers, while SGLT-2 was found in the lung, breast and liver cancers [59][60]. Thus, inhibition of SGLT-2 may have a direct impact on cancer cells growth. Studies in vitro and in animal models documented such effect of dapagliflozin in case of CaKi-1 renal cell cancer line with high expression of SGLT-2 [61], canagliflozin in two cell lines of hepatocellular cancer [62], ipragliflozin, canagliflozin and dapagliflozin in breast cancer [63][64].
The metabolic effect associated with SGLT-2 inhibitor use include lowering of blood glucose level, weight reduction, mainly fat mass, and decreased endogenous insulin secretion or decreased exogenous insulin demand and improved peripheral insulin sensitivity, i.e., they have a positive impact on factors associated with cancer risk. In addition, positive effect associated with SGLT-2 inhibitors use is observed in nonalcoholic fatty liver disease (NAFLD), which is considered as a risk factor of cirrhosis and hepatocellular carcinoma [43]. Thus, SGLT-2 inhibitors can also act on cancer cells indirectly. Jojima et al. demonstrated significantly lower steatosis score in mice treated with canagliflozin compared to vehicle and, in long-term observation, significantly fewer hepatic tumors in the group using continuously canagliflozin compared to the vehicle group. Moreover, canagliflozin in vitro attenuated HepG2 cells proliferation via activation of caspase-3 [65]. Canagliflozin also appeared to be effective in inhibiting lung and prostate cancer cells growth in vitro via inhibiting mitochondrial complex-I supported respiration [66]. In the mouse models of obesity-related cancers: breast and colon cancer, canagliflozin slowed tumor growth. This effect was abrogated by insulin infusion. Thus, in case of obesity-related cancers canagliflosin exerts its oncoprotective effect through insulin- and glucose-lowering action [67]. In a study by Shiba et al. canagliflozin attenuated development of HCC in a mouse model of human NASH. Administration of canagliflozin was associated with improvement in hyperglycemia, hyperinsulinemia, inflammation in liver and adipose tissue, liver steatosis and reactive oxygen species generation, which led to decreased risk of HCC development [68]. Saito et al. demonstrated significant reduction in a number of HCT116 colon cancer cells associated with dapagliflozin treatment, and this effect was independent of SGLT-2 inhibition [69]. Kato et al. revealed in diabetic, obese mice significant suppression of the development of colorectal neoplastic lesions after administration of tofogliflozin. This effect was not associated with direct inhibition of SGLT-2, but with glucose-lowering effect and amelioration of chronic inflammation [70].
On the other hand, also increased risk of certain cancers during the use of SGLT-2 inhibitor was described. Rats treated for two years with canagliflozin at dose 100 mg/kg. showed increased incidence of pheochromocytomas and renal tubular tumors, while testicular Leydig cell tumors were observed also with doses 10 and 30 mg/kg [71]. However, further study revealed that these tumors were secondary to malabsorption of glucose, and were not due to a direct effect of canagliflozin [72]. Moreover, the highest dose of canagliflozin was over 20-fold higher than it is used in clinical practice.
Long-term data from human studies assessing impact of SGLT-2 inhibitors on cancer are scarce. In a population-based cohort study conducted by Suissa et al. with the use of the U.K. Clinical Practice Research Datalink (CPRD), no difference in breast cancer incidence between new SGLT-2 and DPP-4 inhibitors users was found. However, median follow-up was only 2.6 years, thus, SGLT-2 inhibitors might not exert beneficial effects associated with metabolic changes associated with their use [73]. García et al. analyzed association of SGLT-2 inhibitors use and bladder cancer reported in the European Pharmacovigilance Database. They found significantly higher number of cases associated with SGLT-2 inhibitors use compared to other antidiabetic medications, after exclusion of pioglitazone [74]. However, this can be the case similar to association of sitagliptin and exenatide with pancreatic cancer reported by Elashoff M et al. in 2011 [35], which was not confirmed in further observations.
References
- Buse, J.B.; Wexler, D.J.; Tsapas, A.; Rossing, P.; Mingrone, G.; Mathieu, C.; D’Alessio, D.A.; Davies, M.J. 2019 Update to: Management of hyperglycemia in Type 2 diabetes, a consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2020, 43, 487–493.
- Araszkiewicz, A.; Bandurska-Stankiewicz, E.; Budzyński, A.; Cypryk, K.; Czech, A.; Czupryniak, L.; Drzewoski, J.; Dzida, G.; Dziedzic, T.; Zozulińska-Ziółkiewicz, D.; et al. 2020 Guidelines on the management of diabetic patients. A position of Diabetes Poland. Clin. Diabetologia 2020, 9, 1–101.
- Chou, R.; Helfand, M. Challenges in systematic reviews that assess treatment harms. Ann. Intern. Med. 2005, 142, 1090–1099.
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305.
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with Type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258.
- Dąbrowski, M.; Szymańska-Garbacz, E.; Miszczyszyn, Z.; Dereziński, T.; Czupryniak, L. Risk factors for cancer development in type 2 diabetes: A retrospective case-control study. BMC Cancer 2016, 16, 785.
- Dąbrowski, M.; Szymańska-Garbacz, E.; Miszczyszyn, Z.; Dereziński, T.; Czupryniak, L. Differences in risk factors of malignancy between men and women with type 2 diabetes: A retrospective case-control study. Oncotarget 2017, 8, 66940–66950.
- Heckman-Stoddard, B.M.; DeCensi, A.; Sahasrabuddhe, V.V.; Ford, L.G. Repurposing metformin for the prevention of cancer and cancer recurrence. Diabetologia 2017, 60, 1639–1647.
- Schulten, H.-J. Pleiotropic effects of metformin on cancer. Int. J. Mol. Sci. 2018, 19, 2850.
- Wu, T.; Horowitz, M.; Rayner, C.K. New insights into the anti-diabetic actions of metformin: From the liver to the gut. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 157–166.
- Lu, C.-C.; Chiang, J.-H.; Tsai, F.-J.; Hsu, Y.-M.; Juan, Y.-N.; Yang, J.; Chiu, H. Metformin triggers the intrinsic apoptotic response in human AGS gastric adenocarcinoma cells by activating AMPK and suppressing mTOR/AKT signaling. Int. J. Oncol. 2019, 54, 1271–1281.
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an anticancer agent. Trends Pharm. Sci. 2018, 39, 867–878.
- Sanaki, Y.; Nagata, R.; Kizawa, D.; Léopold, P.; Igaki, T. Hyperinsulinemia drives epithelial tumorigenesis by abrogating cell competition. Dev. Cell 2020, 53, 379–389.
- Faria, J.; Negalha, G.; Azevedo, A.; Martel, F. Metformin and breast cancer: Molecular targets. J. Mammary Gland. Biol. Neoplasia 2019, 24, 111–123.
- Xue, J.; Li, L.; Li, N.; Li, F.; Qin, X.; Li, T.; Liu, M. Metformin suppresses cancer cell growth in endometrial carcinoma by inhibiting PD-Leur. J. Pharm. 2019, 859, 172541.
- Kamarudin, M.N.A.; Sarker, M.M.R.; Zhou, J.R.; Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 2019, 38, 491.
- Kawakita, E.; Yang, F.; Kumagai, A.; Takagaki, Y.; Kitada, M.; Yoshitomi, Y.; Ikeda, T.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; et al. Metformin Mitigates DPP-4 Inhibitor-Induced Breast Cancer Metastasis via Suppression of mTOR Signaling. Mol. Cancer Res. 2021, 19, 61–73.
- Seino, S.; Takahashi, H.; Takahashi, T.; Shibasaki, T. Treating diabetes today: A matter of selectivity of sulphonylureas. Diabetes Obes. Metab. 2012, 14, 9–13.
- Currie, C.J.; Poole, C.D.; Gale, E.A. The influence of glucose lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009, 52, 1766–1777.
- Monami, M.; Lamanna, C.; Balzi, D.; Marchionni, N.; Mannucci, E. Sulphonylureas and cancer: A case-control study. Acta Diabetol. 2009, 46, 279–284.
- Chen, Y.; Du, L.; Li, L.; Ma, J.; Geng, X.; Yao, X.; Liu, G.; Sun, X. Cancer risk of sulfonylureas in patients with type 2 diabetes mellitus: A systematic review. J. Diabetes 2017, 9, 482–494.
- Yang, Y.X.; Hennessy, S.; Lewis, J.D. Insulin therapy and colorectal cancer risk among type 2 diabetes mellitus patients. Gastroenterology 2004, 127, 1044–1050.
- Hemkens, L.G.; Grouven, U.; Bender, R.; Günster, C.; Gutschmidt, S.; Selke, G.W.; Sawicki, P.T. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: A cohort study. Diabetologia. 2009, 52, 1732–1744.
- Karlstad, Ø.; Starup-Linde, J.; Vestergaard, P.; Hjellvik, V.; Bazelier, M.T.; Schmidt, M.K.; Andersen, M.; Auvinen, A.; Haukka, J.; Furu, K.; et al. Use of insulin and insulin analogs and risk of cancer: Systematic review and meta-analysis of observational studies. Curr. Drug. Saf. 2013, 8, 333–348.
- Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Schernthaner, G.; Currie, C.J. Glucose-lowering with exogenous insulin monotherapy in type 2 diabetes: Dose association with all-cause mortality, cardiovascular events and cancer. Diabetes Obes. Metab. 2015, 17, 350–362.
- Dąbrowski, M.; Szymańska-Garbacz, E.; Miszczyszyn, Z.; Dereziński, T.; Czupryniak, L. Antidiabetic medications use and cancer risk in type 2 diabetes. Clin. Diabetol. 2017, 6, 17–25.
- The ORIGIN Trial Investigators. Basal insulin and cardiovascular and other outcomes in dysglycemia. N. Engl. J. Med. 2012, 367, 319–328.
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARγ signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85.
- Wu, L.; Zhu, J.; Prokop, L.J.; Murad, M.H. Pharmacologic therapy of diabetes and overall cancer risk and mortality: A meta-analysis of 265 studies. Sci. Rep. 2015, 15, 10147.
- Okumura, T. Mechanisms by which thiazolidinediones induce anti-cancer effects in cancers in digestive organs. J. Gastroenterol. 2010, 45, 1097–1102.
- Tang, H.; Shi, W.; Fu, S.; Wang, T.; Zhai, S.; Song, Y.; Han, J. Pioglitazone and bladder cancer risk: A systematic review and meta-analysis. Cancer Med. 2018, 7, 1070–1080.
- Chen, X.W.; He, Z.X.; Zhou, Z.W.; Yang, T.; Zhang, X.; Yang, Y.X.; Duan, W.; Zhou, S.F. Clinical pharmacology of dipeptidyl peptidase 4 inhibitors indicated for the treatment of type 2 diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2015, 42, 999–1024.
- Capuano, A.; Sportiello, L.; Maiorino, M.I.; Rossi, F.; Giugliano, D.; Esposito, K. Dipeptidyl peptidase-4 inhibitors in Type 2 diabetes therapy-focus on alogliptin. Drug. Des. Dev. Ther. 2013, 7, 989–1001.
- Yang, F.; Takagaki, Y.; Yoshitomi, Y.; Ikeda, T.; Li, J.; Kitada, M.; Kumagai, A.; Kawakita, E.; Shi, S.; Kanasaki, K.; et al. Inhibition of dipeptidyl peptidase-4 accelerates epithelial-mesenchymal transition and breast cancer metastasis via the CXCL12/CXCR4/mTOR Axis. Cancer Res. 2019, 79, 735–746.
- Elashoff, M.; Matveyenko, A.V.; Gier, B.; Elashoff, R.; Butler, P.C. Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 2011, 141, 150–156.
- Overbeek, J.A.; Bakker, M.; van der Heijden, A.A.W.A.; van Herk-Sukel, M.P.P.; Herings, R.M.C.; Nijpels, G. Risk of dipeptidyl peptidase-4 (DPP-4) inhibitors on site-specific cancer: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2018, 34, e3004.
- Dicembrini, I.; Nreu, B.; Montereggi, C.; Mannucci, E.; Monami, M. Risk of cancer in patients treated with dipeptidyl peptidase-4 inhibitors: An extensive meta-analysis of randomized controlled trials. Acta Diabetol. 2020, 57, 689–696.
- Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 2007, 113, 546–593.
- Yabe, D.; Seino, Y. Two incretin hormones GLP-1 and GIP: Comparison of their actions in insulin secretion and β cell preservation. Prog. Biophys. Mol. Biol. 2011, 107, 248–256.
- Liu, Z.; Habener, J.F. Glucagon-like peptide-1 activation of TCF7L2-dependent Wnt signaling enhances pancreatic beta cell proliferation. J. Biol. Chem. 2008, 283, 8723–8735.
- Cao, C.; Yang, S.; Zhou, Z. GLP-1 receptor agonists and risk of cancer in Type 2 diabetes: An updated meta-analysis of randomized controlled trials. Endocrine 2019, 66, 157–165.
- Wright, E.M.; Hirayama, B.A.; Loo, D.F. Active sugar transport in health and disease. J. Intern. Med. 2007, 261, 32–43.
- Simes, B.C.; MacGregor, G.G. Sodium-glucose cotransporter-2 (SGLT2) inhibitors: A clinician’s guide. Diabetes Metab. Syndr. Obes. 2019, 12, 2125–2136.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empa-reg outcome investigators. Empagliflozin, cardiovascular outcomes, and mortality in Type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. CANVAS program collaborative group. Canagliflozin and cardiovascular and renal events in Type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657.
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Declare–timi 58 investigators. Dapagliflozin and cardiovascular outcomes in Type 2 diabetes. N. Engl. J. Med. 2019, 380, 347–357.
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empa-reg outcome Investigators. Empagliflozin and progression of kidney disease in Type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334.
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. CREDENCE trial investigators. Canagliflozin and renal outcomes in Type 2 diabetes and nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306.
- Mosenzon, O.; Wiviott, S.D.; Cahn, A.; Rozenberg, A.; Yanuv, I.; Goodrich, E.L.; Murphy, S.A.; Heerspink, H.J.L.; Zelniker, T.A.; Dwyer, J.P.; et al. Effects of dapagliflozin on development and progression of kidney disease in patients with Type 2 diabetes: An analysis from the DECLARE-TIMI 58 randomised trial. Lancet Diabetes Endocrinol. 2019, 7, 606–617.
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. DAPA-HF trial committees and investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 2019, 381, 1995–2008.
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. EMPEROR-reduced trial investigators. Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 2020, 383, 1413–1424.
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. DAPA-CKD trial committees and investigators. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 2020, 383, 1436–1446.
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. VERTIS CV investigators. Cardiovascular outcomes with ertugliflozin in Type 2 diabetes. N. Engl. J. Med. 2020, 383, 1425–1435.
- Drugs.com. FDA Advisory Committee Makes Recommendation on Investigational Compound Dapagliflozin. Posted 19th July 2011. Available online: (accessed on 5 February 2021).
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium–glucose cotransporter 2 inhibitors for Type 2 diabetes. Ann. Intern. Med. 2013, 159, 262–274.
- Tang, H.; Dai, Q.; Shi, W.; Zhai, S.; Song, Y.; Han, J. SGLT2 inhibitors and risk of cancer in Type 2 diabetes: A systematic review and meta-analysis of randomised controlled trials. Diabetologia 2017, 60, 1862–1872.
- Dicembrini, I.; Nreu, B.; Mannucci, E.; Monami, M. Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: A meta-analysis of randomized controlled trials. Diabetes Obes. Metab. 2019, 21, 1871–1877.
- Bhatt, D.L.; Szarek, M.; Pitt, B.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Inzucchi, S.E.; Kosiborod, M.N.; et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N. Engl. J. Med. 2021, 384, 129–139.
- Madunić, I.V.; Madunić, J.; Breljak, D.; Karaica, D.; Sabolić, I. Sodium-glucose cotransporters: New targets of cancer therapy? Arch. Ind. Hyg. Toxicol. 2018, 69, 278–285.
- Perry, R.J.; Shulman, G.I. Sodium-glucose cotransporter-2 inhibitors: Understanding the mechanisms for therapeutic promise and persisting risks. J. Biol. Chem. 2020, 295, 14379–14390.
- Kuang, H.; Liao, L.; Chen, H.; Kang, Q.; Shu, X.; Wang, Y. Therapeutic effect of sodium glucose co-transporter 2 inhibitor dapagliflozin on renal cell carcinoma. Med. Sci. Monit. 2017, 23, 3737–3745.
- Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722.
- Komatsu, S.; Nomiyama, T.; Numata, T.; Kawanami, T.; Hamaguchi, Y.; Iwaya, C.; Horikawa, T.; Fujimura-Tanaka, Y.; Hamanoue, N.; Motonaga, R.; et al. SGLT2 inhibitor ipragliflozin attenuates breast cancer cell proliferation. Endocr. J. 2020, 67, 99–106.
- Zhou, J.; Zhu, J.; Yu, S.-J.; Ma, H.-L.; Chen, J.; Ding, X.-F.; Chen, G.; Liang, Y.; Zhang, Q. Sodium-glucose co-transporter-2 (SGLT-2) inhibition reduces glucose uptake to induce breast cancer cell growth arrest through AMPK/mTOR pathway. Biomed. Pharm. 2020, 132, 110821.
- Jojima, T.; Wakamatsu, S.; Kase, M.; Iijima, T.; Maejima, Y.; Shimomura, K.; Kogai, T.; Tomaru, T.; Usui, I.; Aso, Y.; et al. The SGLT2 inhibitor canagliflozin prevents carcinogenesis in a mouse model of diabetes and non-alcoholic steatohepatitis-related hepatocarcinogenesis: Association with SGLT2 expression in hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 5237.
- Villani, L.A.; Smith, B.K.; Marcinko, K.; Ford, R.J.; Broadfield, L.A.; Green, A.E.; Houde, V.P.; Muti, P.; Tsakiridis, T.; Steinberg, G.R. The diabetes medication Canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol. Metab. 2016, 5, 1048–1056.
- Nasiri, A.R.; Rodrigues, M.R.; Li, Z.; Leitner, B.P.; Perry, R.J. SGLT2 inhibition slows tumor growth in mice by reversing hyperinsulinemia. Cancer Metab. 2019, 7, 1–13.
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362.
- Saito, T.; Okada, S.; Yamada, E.; Shimoda, Y.; Osaki, A.; Tagaya, Y.; Shibusawa, R.; Okada, J.; Yamada, M. Effect of dapagliflozin on colon cancer cell Rapid Communication. Endocr. J. 2015, 62, 1133–1137.
- Kato, J.; Shirakami, Y.; Ohnishi, M.; Mizutani, T.; Kubota, M.; Sakai, H.; Ibuka, T.; Tanaka, T.; Shimizu, M. Suppressive effects of the sodium-glucose cotransporter 2 inhibitor tofogliflozin on colorectal tumorigenesis in diabetic and obese mice. Oncol. Rep. 2019, 42, 2797–2805.
- De Jonghe, S.; Proctor, J.; Vinken, P.; Feyen, B.; Wynant, I.; Marien, D.; Geys, H.; Mamidi, R.N.; Johnson, M.D. Carcinogenicity in rats of the SGLT2 inhibitor canagliflozin. Chem. Interact. 2014, 224, 1–12.
- De Jonghe, S.; Johnson, M.D.; Mamidi, R.N.; Vinken, P.; Feyen, B.; Lammens, G.; Proctor, J. Renal tubular and adrenal medullary tumors in the 2-year rat study with canagliflozin confirmed to be secondary to carbohydrate (glucose) malabsorption in the 15-month mechanistic rat study. Chem. Interact. 2017, 277, 85–90.
- Suissa, M.; Yin, H.; Yu, O.H.; Wong, S.M.; Azoulay, L. Sodium–Glucose cotransporter 2 inhibitors and the short-term risk of breast cancer among women with Type 2 Diabetes. Diabetes Care 2021, 44, e9–e11.
- García, M.; Arteche-Martinez, U.; Lertxundi, U.; Aguirre, C. SGLT2 inhibitors and bladder cancer: Analysis of cases reported in the European pharmacovigilance database. J. Clin. Pharm. 2021, 61, 187–192.