Label-free microscopy methods rely on photophysical processes to generate signals through specific interactions with biological molecules and offer great potential for basic research and clinical applications.
- multiphoton microscopy
- label-free
- second harmonic generation
1. Introduction
Light microscopy is a gold standard technique in biomedical research and clinical diagnosis [1]. The huge technical developments toward more and more sophisticated apparatus [2][3][4] enhanced the broad use of light microscopy, which in turn increased the need for innovative microscopy approaches. Some examples of these multidisciplinary solutions are nonlinear optical microscopy, super-resolution [5][6][7], fluorescent markers, and optimization of sample preparation [8][9]. Confocal laser scanning microscopy is routinely used in biomedical research because it ensures high resolution and high contrast compared to epifluorescence microscopes (for a complete review, see Jonkman et al. [10]). A drawback of this technique is the limited penetration into the sample (50–100 µm) and the photodamage caused by illumination light, which is usually in the range of 400–600 nm. Thanks to the use of longer wavelengths (800–1200 nm), multiphoton microscopy reduces sample damage and provides deeper penetration (250–500 µm) into the specimen, although with some loss of resolution. This technique has found success as a non-invasive imaging tool for thick biological tissues and living animals. There are typically two labeling strategies for multiphoton experiments. Fluorescent proteins may be expressed under genetic control throughout a tissue sample; however, this requires complex and time-consuming genetic manipulation of the model organism genome. Alternatively, conventional antibody labeling strategies may be employed; however, the penetration of probes into tissue is not straightforward and may require heavy detergent action, which can disrupt tissue ultra-structure. Of notice, any external operation may alter the intrinsic characteristic of the specimen. For this reason, the possibility to analyse biological samples without labeling procedures while maintaining molecular specificity is becoming more and more popular.
Light microscopy is a gold standard technique in biomedical research and clinical diagnosis [1]. The huge technical developments toward more and more sophisticated apparatus [2,3,4] enhanced the broad use of light microscopy, which in turn increased the need for innovative microscopy approaches. Some examples of these multidisciplinary solutions are nonlinear optical microscopy, super-resolution [5,6,7], fluorescent markers, and optimization of sample preparation [8,9]. Confocal laser scanning microscopy is routinely used in biomedical research because it ensures high resolution and high contrast compared to epifluorescence microscopes (for a complete review, see Jonkman et al. [10]). A drawback of this technique is the limited penetration into the sample (50–100 µm) and the photodamage caused by illumination light, which is usually in the range of 400–600 nm. Thanks to the use of longer wavelengths (800–1200 nm), multiphoton microscopy reduces sample damage and provides deeper penetration (250–500 µm) into the specimen, although with some loss of resolution. This technique has found success as a non-invasive imaging tool for thick biological tissues and living animals. There are typically two labeling strategies for multiphoton experiments. Fluorescent proteins may be expressed under genetic control throughout a tissue sample; however, this requires complex and time-consuming genetic manipulation of the model organism genome. Alternatively, conventional antibody labeling strategies may be employed; however, the penetration of probes into tissue is not straightforward and may require heavy detergent action, which can disrupt tissue ultra-structure. Of notice, any external operation may alter the intrinsic characteristic of the specimen. For this reason, the possibility to analyse biological samples without labeling procedures while maintaining molecular specificity is becoming more and more popular.
Label-free microscopy methods rely on photophysical processes to generate signals through specific interactions with biological molecules and offer great potential for basic research and clinical applications. Multiphoton microscopy is probably the most popular label-free technique. Using the same optical path, it can identify three different types of signal: autofluorescence, second harmonic generation and third harmonic generation. Two-photon autofluorescence has been widely used to identify and quantify metabolic molecules such as NADH and tryptophan [11][12][13]. Using fluorescence lifetime imaging (FLIM), it is even possible to quantify the proportion of NADH that is free or protein-bound [14] and distinguish NADH from NADPH [15][16][17]. In contrast, second and third harmonic signals (SHG and THG) are non-fluorescent photo-physical conversions dependent on the intrinsic properties of the target biomaterial [7][18]. A variety of molecules have been reported to generate second harmonic signals, such as collagen, myosin, microtubules, silk, starch and cellulose. All these molecules are characterized by non-centrosymmetric architecture or hyperpolarizability, making them SHG active molecules (also named harmonophores) [19][20]. In SHG microscopy, two photons of the same frequency pass through these biomaterials and result in a photon with doubled frequency (and thus half wavelength). Otherwise, third harmonic generation requires the presence of an interface characterized by remarkably different refraction indexes that cause a symmetry break or by molecules with third order nonlinear susceptibility. Some examples of THG sources are lipid droplets, elastin fibers, bone calcifications, and cellular membranes [21].
Label-free microscopy methods rely on photophysical processes to generate signals through specific interactions with biological molecules and offer great potential for basic research and clinical applications. Multiphoton microscopy is probably the most popular label-free technique. Using the same optical path, it can identify three different types of signal: autofluorescence, second harmonic generation and third harmonic generation. Two-photon autofluorescence has been widely used to identify and quantify metabolic molecules such as NADH and tryptophan [11,12,13]. Using fluorescence lifetime imaging (FLIM), it is even possible to quantify the proportion of NADH that is free or protein-bound [14] and distinguish NADH from NADPH [15,16,17]. In contrast, second and third harmonic signals (SHG and THG) are non-fluorescent photo-physical conversions dependent on the intrinsic properties of the target biomaterial [7,18]. A variety of molecules have been reported to generate second harmonic signals, such as collagen, myosin, microtubules, silk, starch and cellulose. All these molecules are characterized by non-centrosymmetric architecture or hyperpolarizability, making them SHG active molecules (also named harmonophores) [19,20]. In SHG microscopy, two photons of the same frequency pass through these biomaterials and result in a photon with doubled frequency (and thus half wavelength). Otherwise, third harmonic generation requires the presence of an interface characterized by remarkably different refraction indexes that cause a symmetry break or by molecules with third order nonlinear susceptibility. Some examples of THG sources are lipid droplets, elastin fibers, bone calcifications, and cellular membranes [21].
A comparison between different microscopy approaches can vary depending on the specific application (
). For the analysis of structural components within tissues, label-free multiphoton (LFM) microscopy can be successfully used to retrieve information in a staining-free fashion. This results in a great advantage in terms of sample preparation ease where the absence of immunofluorescence or fluorescent protein expression simplifies benchwork protocols. On the other hand, despite the high signal specificity obtained by SHG and THG, the molecular species that can be monitored in LFM are limited. In multiphoton fluorescence (MPF) and, even more, in confocal fluorescence microscopy, a huge number of antibodies and fluorescent tags have been developed to mark the protein of interest, overcoming this limitation. Confocal microscopy, based on visible wavelengths, possesses higher resolution compared to MPF and LFM that use longer excitation wavelengths. Moreover, LFM is more sensitive than MPF to a degradation of the focal spot caused by scattering inside the sample that reduces conversion efficiency [22]. For the same reason, LFM depth penetration is slightly reduced compared to MPF on the same sample. Lastly, sample integrity can be estimated in terms of photobleaching and phototoxicity during imaging. In LFM, photobleaching does not occur for SHG and THG molecules, while it has to be considered for fluorescent dyes and proteins.
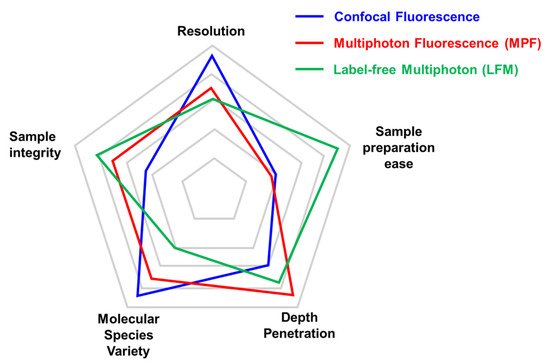
Figure 1.
Comparison of key parameters of different imaging techniques. Each microscopy technique has strengths and weaknesses that are important to consider before choosing the adequate microscopy approach. We limited the radar plot to five parameters (Resolution, Sample preparation ease, Depth penetration, Molecular species variety, and Sample integrity), although experimental needs may require considering other aspects (i.e., acquisition speed). The relative performance of confocal microscopy (in blue), multiphoton (in red), and label-free multiphoton (in green) are compared with the outer position indicating the best performance for that parameter. The same set-up can be optimized for specific approaches (multiphoton fluorescence vs. label-free multiphoton).
Other label-free imaging techniques have been developed and are based on a variety of optical approaches. Holotomographic microscopy and spiral phase microscopy are sensitive to variations of the refractive index of cellular compartments and have been used to study organelle dynamics [23] and enhance edge contrast [24]. Raman microscopy can identify chemical bonds and is particularly interesting to quantify lipids abundance. Variants of Raman microscopy include CARS (Coherent Anti-Stokes Raman Spectroscopy) and SRS (Stimulated Raman Spectroscopy). Both achieve a better signal to noise ratio than Raman microscopy and are amenable to imaging small molecules that are not easy to tag with a fluorophore. Photoacoustic imaging exploits the conversion of incident laser light into heat by the tissue to reconstruct an image [25][26]. The optical resolution of this technique is low compared to optical microscopy; however, it achieves deeper penetration (several millimeters). Lastly, interferometric scattering (iSCAT) microscopy is a light scattering technique that is sensitive and can be used to determine the molecular weight of single unlabeled proteins released by living cells [27][28].
Other label-free imaging techniques have been developed and are based on a variety of optical approaches. Holotomographic microscopy and spiral phase microscopy are sensitive to variations of the refractive index of cellular compartments and have been used to study organelle dynamics [23] and enhance edge contrast [24]. Raman microscopy can identify chemical bonds and is particularly interesting to quantify lipids abundance. Variants of Raman microscopy include CARS (Coherent Anti-Stokes Raman Spectroscopy) and SRS (Stimulated Raman Spectroscopy). Both achieve a better signal to noise ratio than Raman microscopy and are amenable to imaging small molecules that are not easy to tag with a fluorophore. Photoacoustic imaging exploits the conversion of incident laser light into heat by the tissue to reconstruct an image [25,26]. The optical resolution of this technique is low compared to optical microscopy; however, it achieves deeper penetration (several millimeters). Lastly, interferometric scattering (iSCAT) microscopy is a light scattering technique that is sensitive and can be used to determine the molecular weight of single unlabeled proteins released by living cells [27,28].
2. Multiphoton Technique
Multiphoton microscopy (MPM) encompasses several laser-scanning methods based on the nonlinear interaction of light with the specimen. In this context, “nonlinear” means that the intensity of the signal depends on the simultaneous interaction of the probe with two or three photons [29]. In single photon microscopy, photons at a determined wavelength can excite the target fluorophore, delivering the adequate amount of energy to induce the transition to the excited state (S1). Then, the fluorophore relaxes back to the ground state (S0) emitting a photon of reduced energy and longer wavelength. In MPM, the use of longer wavelengths comes with photons of halved energy (for two-photon microscopy). For this reason, to obtain the excitation of the fluorophore to the S1 state, two exciting photons are needed. The simultaneous interaction with more than one photon requires an extremely high density of photons at the focal point of the objective, which is typically achieved using pulsed lasers producing mega-watts to giga-watts of power. The optical sectioning capability of MPM results from the selective generation of a signal in the focal plane of the objective. This approach enables the collection of all emitted photons, even the scattered ones, without the need for pinhole filtering used in confocal microscopy, since all signals are generated in the focal volume [1]. Moreover, the longer wavelengths used in MPM face less scattering and absorption by biological matter, allowing reaching deeper layers inside the sample. Therefore, MPM represents the best non-invasive technique to achieve imaging in deep explanted tissues or living animals [1][30]. The advantage of the volume-confined excitation is that the lack of out-of-focus excitation in two-photon excitation (TPE) reduces specimen photobleaching in non-imaged areas. This also means that photodamage is highly confined, allowing long-term observations of biological specimens that would otherwise be limited using single photon excitation. Of notice, in the focal volume, the illumination power must be controlled carefully. Photobleaching and photodamage are also nonlinear phenomena where a higher order of photon absorption has been observed [31].
Multiphoton microscopy (MPM) encompasses several laser-scanning methods based on the nonlinear interaction of light with the specimen. In this context, “nonlinear” means that the intensity of the signal depends on the simultaneous interaction of the probe with two or three photons [29]. In single photon microscopy, photons at a determined wavelength can excite the target fluorophore, delivering the adequate amount of energy to induce the transition to the excited state (S1). Then, the fluorophore relaxes back to the ground state (S0) emitting a photon of reduced energy and longer wavelength. In MPM, the use of longer wavelengths comes with photons of halved energy (for two-photon microscopy). For this reason, to obtain the excitation of the fluorophore to the S1 state, two exciting photons are needed. The simultaneous interaction with more than one photon requires an extremely high density of photons at the focal point of the objective, which is typically achieved using pulsed lasers producing mega-watts to giga-watts of power. The optical sectioning capability of MPM results from the selective generation of a signal in the focal plane of the objective. This approach enables the collection of all emitted photons, even the scattered ones, without the need for pinhole filtering used in confocal microscopy, since all signals are generated in the focal volume [1]. Moreover, the longer wavelengths used in MPM face less scattering and absorption by biological matter, allowing reaching deeper layers inside the sample. Therefore, MPM represents the best non-invasive technique to achieve imaging in deep explanted tissues or living animals [1,30]. The advantage of the volume-confined excitation is that the lack of out-of-focus excitation in two-photon excitation (TPE) reduces specimen photobleaching in non-imaged areas. This also means that photodamage is highly confined, allowing long-term observations of biological specimens that would otherwise be limited using single photon excitation. Of notice, in the focal volume, the illumination power must be controlled carefully. Photobleaching and photodamage are also nonlinear phenomena where a higher order of photon absorption has been observed [31].
Thanks to its high-resolution capabilities, MPM has provided unprecedented possibilities for the study of neuroscience [32], metastasis [33], calcium imaging [6], embryonic development, and cell–cell in vivo interactions.
The most common MPM variation is two-photon excitation (TPE) microscopy [30] (
a). This technique is based on the simultaneous adsorption of two photons by a fluorophore in a single quantum event. This phenomenon was theoretically predicted by Maria Goeppert-Mayer in 1931 but could be experimentally verified only after the advent of lasers. In 1961, Peter Franken and colleagues demonstrated the frequency doubling of light focusing a ruby laser on a quartz crystal [34]. The development of ultrashort pulsed Ti:Sapphire mode-locked lasers allowed more practical generation of the necessary photon density. Exploitation of two-photon laser excitation laid dormant until Denk et al. devised a practicable two-photon laser-scanning fluorescence microscope [30] that has opened new frontiers in biomedical research.
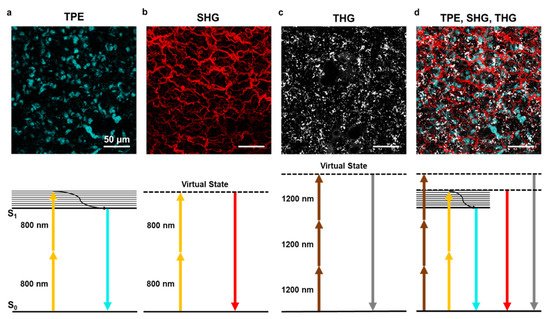
Figure 2.
Two-photon excitation, second and third harmonic generation. (
a
) Example of two-photon excitation (TPE) of DAPI stained nuclei in lung tissue. Bottom panel shows the corresponding Jablonski diagram using 800 nm excitation wavelength. (
b
) Second harmonic generation (SHG) signal elicited with 800 nm wavelength on lung tissue collagen structure. The bottom panel shows the corresponding Jablonski diagram using 800 nm excitation wavelength. (
c
) Third harmonic generation (THG) signal of lipid bodies in lung tissue using 1200 nm wavelength. The bottom panel shows the corresponding Jablonski diagram. (
d
) A combination of TPE, SHG, and THG can be obtained using two laser sources at different wavelengths. Images obtained with the 800 nm source are merged with THG obtained with the 1200 nm source. The scale bar is 50 µm for all images.
The occurrence of the double-absorption event is determined by photon–molecule interactions, which must occur with a relative delay shorter than the typical virtual state lifetime of a given fluorophore [30]. Furthermore, the two-photon excitation rate depends on the second power of the incident light intensity and is ≈10–14 times smaller than the one-photon absorption rate; hence, the successful implementation of TPE imaging requires extremely high photon fluxes, which in practice translates into the use of mode-locked laser sources with pulse durations below 1
ps
and frequencies of about 100 MHz. Ti:Sapphire lasers ensure adequate tunable sources in the 760–960 nm window and are the most used lasers for MPM. In the last years, the commercial availability of laser sources has grown, opening to new wavelength windows beyond the Ti:Sapphire range, with the advent of optical parametric oscillators (OPO) whose typical spectral range is 1030–1300 nm. An additional option is offered by ultrashort pulsed lasers, which can operate at specific wavelengths, such as 775, 1030–1060, and 1500 nm. The combination of these two laser sources allows multimodal excitation of the sample together with wavelength mixing that consists of the simultaneous absorption of two different wavelengths enabling different state transitions. This resulted in an optimal configuration for the excitation of certain fluorophores, such as YFP, whose absorption spectra is centered outside the Ti:Sapphire emission laser [35]. The acquisition wavelengths depend on the setup design but usually range from 380 to 700 nm, ensuring proper separation of excitation and emission signal.
Despite the need for very intense light sources, TPE presents several advantages over classical single-photon techniques: the wavelengths used are in the near-IR, making them less subjected to scattering or absorption from biological thick specimens (which means deeper penetration capabilities) [36]; due to the high photon flux required to achieve the two-photon absorption, the focal volume is very small, about 0.1 μm
3
[37]. Such a small focal volume is the main reason for the inherent ability of MPM to perform axial sectioning of samples and for the photobleaching restricted to the focus plane [38]. The effective spatial confinement is a direct consequence of the insignificant single-photon fluorescence excited in out-of-focus regions. Note that TPE can be used to generate fluorescence from all possible fluorophores, including fluorescent proteins, chemical dyes, and autofluorescence.
The use of different excitation wavelengths combining the Ti:Sapphire laser with an OPO allows multiplexed fluorescence and label-free analysis of the same sample, as shown in
d on a lung ex vivo sample.
3. Label-Free Multiphoton Microscopy in Biomedical Research
A special form of MPM that is becoming more successful in the biomedical context is label-free microscopy. The combined imaging of autofluorescence with second and third harmonic generation (SHG and THG) provides molecular information about the sample without any staining procedure.
3.1. Autofluorescence
The autofluorescence signal is generated by molecular components of cells or matrices and can be used to reconstruct tissue morphology without any staining. Endogenous sources of autofluorescence signals are metabolic substrates (e.g., NADH and FAD), structural proteins (e.g., elastin and keratin), lipofuscins, and melatonin. This means that active cells are perfect sources of autofluorescence signals. A peculiar application of autofluorescence imaging can be found in regenerative medicine. In this biomedical field, tissues from animals or human biopsies are decellularized to constitute ex novo scaffolds for induced pluripotent stem cells (iPS) repopulation or patient-derived 3D models [39][40]. The success of the decellularization procedure is confirmed by DNA quantification as a gold standard reference; however, the presence or absence of cellular components can be assessed by autofluorescence imaging with the advantage that the analyzed tissue remains intact and can be used for other purposes. Moreover, autofluorescence from elastin or other structural proteins gives useful information on scaffold integrity and preservation. On the other hand, autofluorescence signals from metabolic substrates, such as NADH, can be efficiently used to discriminate cellular activity. For example, in muscle biopsies, NADH autofluorescence intensity correlates with the metabolic state of the fibers [7].
The autofluorescence signal is generated by molecular components of cells or matrices and can be used to reconstruct tissue morphology without any staining. Endogenous sources of autofluorescence signals are metabolic substrates (e.g., NADH and FAD), structural proteins (e.g., elastin and keratin), lipofuscins, and melatonin. This means that active cells are perfect sources of autofluorescence signals. A peculiar application of autofluorescence imaging can be found in regenerative medicine. In this biomedical field, tissues from animals or human biopsies are decellularized to constitute ex novo scaffolds for induced pluripotent stem cells (iPS) repopulation or patient-derived 3D models [39,40]. The success of the decellularization procedure is confirmed by DNA quantification as a gold standard reference; however, the presence or absence of cellular components can be assessed by autofluorescence imaging with the advantage that the analyzed tissue remains intact and can be used for other purposes. Moreover, autofluorescence from elastin or other structural proteins gives useful information on scaffold integrity and preservation. On the other hand, autofluorescence signals from metabolic substrates, such as NADH, can be efficiently used to discriminate cellular activity. For example, in muscle biopsies, NADH autofluorescence intensity correlates with the metabolic state of the fibers [7].
3.2. Second Harmonic Generation
Second harmonic generation is a nonlinear coherent light-scattering phenomenon, resulting from the interaction of light waves with molecular structures that have specific crystal-like physical properties. SHG results from the conversion of two incoming photons into one emitted photon having twice the energy and therefore half the wavelength. An SHG signal can be obtained with both Ti:Sapphire and OPO lasers at any wavelength from 800 to 1200 nm. The dependence of SHG signal on incident light intensity must be considered in LFM imaging when using Ti:Sapphire lasers and OPO that display an intensity profile peaked at shorter wavelengths (around 800 nm) and then decline toward longer wavelengths, and it has been demonstrated by Campagnola and colleagues that shorter wavelengths are more efficient in SHG microscopy [22]. The emitted signal is generally collected in transmission due to the momentum and energy conservation properties of the generated SHG signal. Moreover, molecules such as collagen and starch allow collection of the SHG signal both in reflection or transmission. For collagen, this feature has been used to obtain a quantitative evaluation of fibrillar structure [41]. The SHG signal collected in transmission is known as a forward signal, which is more intense than an epicollected signal, which is named backward SHG. SHG allows the identification of molecules with non-centrosymmetric structure via interaction with incident light. In this way, the intrinsic properties of the biological tissue can be studied with no need for contrast-enhancing or labeling while providing high-resolution 3D label-free reconstruction of the imaged portion. The second harmonic generation has been applied to a plethora of samples ranging from zebrafish embryos to bone, fat and skin tissue, brain, heart, and muscles. In all these tissues and organs, some features are particularly efficient at generating second harmonic signals. Of note, collagen is very effective in SHG, resulting in one of the most frequently analyzed matrix components in label-free microscopy (
b). Another SH generator is myosin, which is a key protein in cardiac and muscular tissues. Some protocols to discriminate between the two have been developed and are mainly based on polarization setup.
3.3. Third Harmonic Generation
The third harmonic signal is generated at interfaces and structural inhomogeneities through the interaction with incident light. THG occurs at structural interfaces [21] such as boundaries of regions with highly different refractive indexes. Lipid droplets are particularly efficient THG structures [42] (
c); moreover, elastin fibers, bone calcification, and cellular membrane can be visualized with this technique. While every biological specimen is extremely rich in interfaces, thus being a perfect theoretical candidate for THG, in practice, THG imaging is much less applied and requires a highly specialized multiphoton microscope with long laser wavelengths. THG signals occur at one-third of the illumination wavelength; for example illumination at 900 nm would produce a signal at 300 nm. However, detection in the UV range is compromised by the high absorbance of this wavelength in any biological tissue. Practically, the range 380–450 nm is well suited for THG imaging but requires the availability of longer wavelength lasers (or OPO) in the region above 1050 nm. This ensures minimal absorption and overlap with signals in green and red fluorophores spectral regions, as well as SHG detection at 530–600 nm. Last but not least, THG efficiency is sensitive to laser power, and even a small loss of photons in the focal region may compromise imaging quality. In deep tissues, THG signals are affected by light scattering and aberrations introduced along the optical path within the sample.
3.4. Research and Clinical Applications
The success of label-free multiphoton imaging is well represented by its wide use in many diverse fields from regenerative medicine to cancer and embryogenesis.
Table 1 reports a representative overview of application fields and results obtained thanks to label-free multiphoton microscopy both at the basic research level and clinical diagnosis. Label-free microscopy started as a technically demanding method for experienced users. However, the reduced cost and improved stability of pulsed lasers led to the increased availability of user-friendly systems and the application of nonlinear imaging methods in many biomedical fields. More recently, proof-of-concepts development of the technique is moving from imaging bench to clinical application in optical biopsies. An excellent example of clinical application is DermaInspect, which is a high-resolution multiphoton microendoscopy used in clinical practice to identify melanoma lesions [43][44].
reports a representative overview of application fields and results obtained thanks to label-free multiphoton microscopy both at the basic research level and clinical diagnosis. Label-free microscopy started as a technically demanding method for experienced users. However, the reduced cost and improved stability of pulsed lasers led to the increased availability of user-friendly systems and the application of nonlinear imaging methods in many biomedical fields. More recently, proof-of-concepts development of the technique is moving from imaging bench to clinical application in optical biopsies. An excellent example of clinical application is DermaInspect, which is a high-resolution multiphoton microendoscopy used in clinical practice to identify melanoma lesions [43,44].
Table 1.
Fields of application of label-free multiphoton microscopy.
| Biomedical Field | Representative Results | ||
|---|---|---|---|
| Regenerative medicine and tissue engineering |
| ||
| |||
| Cancer |
| ||
| |||
| |||
| |||
| Cardiovascular |
| ||
| |||
| Neuroscience |
| ||
| |||
| In vitro 3D models |
| ||
| |||
| Development and embryogenesis |
| ||
| |||
| Immunology |
| ||
| |||
| Ophthalmology |
| ||
| Respiratory disease |
| ||
| |||
| Muscle physiology and pathology |
| ||
| |||
| Kidney, Colon, and Liver |
| ||
| |||
|