Titin, also called connectin, is a giant sarcomere protein, which functions as a spring for muscle extension and elasticity. Titin interconnects the contraction of actin-containing thin filaments and myosin-containing thick filaments. Recently, the N-terminal fragment of titin, which is the breakdown product of titin, has become measurable using an enzyme-linked immunosorbent assay kit (27900 titin N-fragment Assay Kit; Immuno-Biological Laboratories, Fujioka, Japan). This kit has been used to evaluate muscle breakdown in muscle dystrophy, in which the level of urinary titin N-fragment was 700-times above the normal level.
- titin
- muscle
1. Titin
Titin, initially known as connectin, was discovered in 1976 by Maruyama et al. [1]. Being the largest protein in the human body, it was renamed titin after the giant god, Titan, from Greek mythology. Titin is the largest protein in humans, and is 3.0–3.7 MDa. This protein is located in the muscle sarcomere, and interconnects the contraction of actin-containing thin filaments and myosin-containing thick filaments. Passive tension and elasticity during muscle contraction develops as a result of the titin protein by Ca
2+
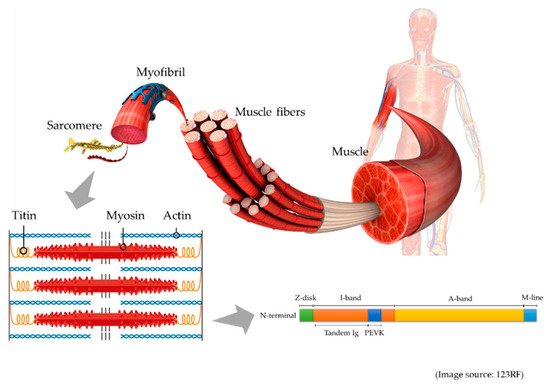
Figure 1.
During muscle degradation, titin is broken down into small fragments, and several different fragments are measurable. In serum, the metalloproteinase (MMP) 2-cleaved titin fragment and MMP 12-cleaved titin fragment are measurable. MMP 2-cleaved titin reflected muscle atrophy in a human bed rest study [4]. On the other hand, MMP 12-cleaved titin fragment could be used to assess cardiac infarction, because the level of the fragment increased after an acute myocardial infarction [5].
Recently, the N-terminal fragment of titin, which is 25 kDa, and is known as the urinary titin N-fragment, has become measurable in urine. Unlike serum, this urinary biomarker is noninvasive, but requires correction by urinary creatinine to adjust for the kidney function. The standard value of urinary titin N-fragment was 2.1 (1.2–2.6) pmol/mg Cr in healthy adult volunteers [6].
2. Muscle Atrophy
2.1. Limb and Trunk Muscle Atrophy
Muscle atrophy is a serious problem in critically ill patients [7]. After one week of ICU admission, critically ill patients exhibited 13.2–16.9% of muscle atrophy in the upper limbs, and 18.8–20.7% in the lower limbs [8]. These muscle atrophies are associated with impaired physical function and mortality [9][10].
Muscle atrophy is caused by an increase in protein degradation or decrease in protein synthesis. Increased protein degradation occurs mainly due to inflammation and immobilization. Calpain, caspase, ubiquitin-proteasome, and autophagy-lysosome have been implicated in this protein degradation pathway [11][12][13]. Decreased protein synthesis is caused by suppressed insulin-like growth factor-1 and inactivated myogenesis [14].
In critically ill patients, inflammation is an important cause of muscle atrophy [15], and various inflammatory cytokines are associated with muscle atrophy [16]. Immobilization is frequently observed during critical illness, and causes muscle atrophy [17]. Malnutrition causes a decrease in protein synthesis [18]. In critically ill patients, recommended protein intake is 1.2–2.0 g/kg/day [19], but this level of intake is often not achieved in the ICU [20], resulting in decreased protein synthesis.
Muscle mass can be assessed using computed tomography, bioelectrical impedance analysis, and ultrasound [21]. Computed tomography is accurate, but exposes patients to radiation, whereas bioelectrical impedance analysis is influenced by fluid changes in critically ill patients [22]. Ultrasound can be used to monitor muscle atrophy at the bedside, but it requires a skilled and experienced operator [23][24]. Thus, a biomarker to assess muscle atrophy is urgently needed. In a rat study, Udaka et al. found that six weeks of immobilization caused titin loss in the soleus muscle, which was associated with muscle dysfunction [25]. Thus, it is theoretically reasonable to expect levels of urinary titin to be elevated in the urine of patients with muscle atrophy.
p < 0.01) [26]. This result possibly reflects the amount of muscle breakdown in these conditions. Recently, two studies have reported the usefulness of the urinary titin N-fragment in the evaluation of muscle atrophy in critically ill patients. Furthermore, Nakano et al. reported that urinary titin N-fragment could be used to evaluate muscle atrophy in critically ill patients [27]. In their study, investigating four critically ill patients, there was a negative correlation between mean urinary titin level during the first seven days of ICU admission and femoral muscle volume measured using computed tomography (
r
p ≤ 0.03), suggesting that urinary titin reflects muscle atrophy in nonsurgical critically ill patients [28]. However, in their study, the correlation between muscle atrophy and urinary titin was limited to
r
p
p
The molecular mechanism underlying titin breakdown remains unclear. Several pathways, activated by inflammation and immobilization, are involved in the breakdown of titin. Calpain contributes to the cleavage of titin because titin has calpain-binding sites [29]. Lang et al. investigated the ubiquitination of titin in denervated mouse, and found that levels of ubiquitinated titin gradually increased in denervation-induced muscle atrophy [30]. Consistent with this finding, Swist et al. found increased levels of ubiquitinated titin in patients with critical illness [31]. In their study, the markers of the autophagy-lysosome pathway were also upregulated. Thus, the autophagy-lysosome pathway may be involved in the breakdown of titin.
Unlike other promising biomarkers of muscle atrophy, urinary titin N-fragment is noninvasive and reliable. Creatinine kinase and BUN/Cr are possible biomarkers for muscle atrophy [32], but these biomarkers require blood tests. Furthermore, creatinine kinase is derived from various tissues [33], and BUN/Cr is influenced by various conditions including dehydration [32]. Urinary creatinine has also been suggested to be a biomarker of muscle atrophy, but it does not consider kidney function [34]. Thus, urinary titin N-fragment, corrected by urinary creatinine, is a reliable biomarker because it does not depend on kidney function [28][35].
2.2. Diaphragm Muscle Atrophy
Diaphragm atrophy is observed in 60% of mechanically ventilated critically ill patients [36], and it is a serious problem because of its association with prolonged mechanical ventilation and prolonged ICU stay [37]. As with limb muscle atrophy, diaphragm atrophy is caused by the calpain, caspase, ubiquitin-proteasome, and autophagy-lysosome pathways [38][39][40][41]. As reported in limb muscles, inflammation and immobilization are important causes of diaphragm atrophy. Sepsis is a cause of diaphragm atrophy [42], and deep sedation causes immobilization of the diaphragm [43]. Most importantly, ventilator settings strongly influence diaphragm atrophy and subsequent diaphragm dysfunction. Thus, diaphragm dysfunction in such cases is termed as ventilator induced diaphragm dysfunction [44].
Titin plays an important role in the diaphragm contractile force [45], and titin loss has been associated with diaphragm dysfunction in rats [46][47]. In the diaphragm biopsy of human subjects, Hussain et al. found that prolonged controlled mechanical ventilation decreased titin levels and impaired the diaphragm myofibrillar force [48]. Furthermore, Lindqvist et al. suggested that the positive-end expiratory pressure (PEEP) during ventilation led to the breakdown of the diaphragm titin, because the PEEP stretched out the sarcomere of the diaphragm muscle fibers [49]. Excessive extension may be detrimental to diaphragm titin.
p = 0.33) [28]. The urinary titin N-fragment can measure the titin breakdown product in all muscles, and is not specific to the diaphragm. To quantify the diaphragm atrophy, a diaphragm-specific titin kit is necessary. This may be theoretically possible, because a cardiac-specific titin kit has been developed in another study [50].
Interestingly, several studies have reported that, as well as diaphragm atrophy, increased diaphragm thickness has worsened clinical outcomes [36][37][51]. Insufficient ventilatory support leads to excessive respiratory effort in mechanically ventilated patients. This condition increases the diaphragm thickness. Since the increased muscle thickness has worsened outcomes, the hypertrophied diaphragm may not have sufficient functional titin to function appropriately. In a previous study on urinary titin N-fragment, there was no significant difference in the cumulative urinary titin N-fragment between the unchanged diaphragm thickness and increased diaphragm thickness groups (147.9 (79.0–257.8) vs. 426.1 (140.8–578.2) pmol/mg Cr in unchanged vs. increased,
p = 0.45) [28]. The combination of increased diaphragm thickness and atrophy also did not have a significant difference in terms of the cumulative level of urinary titin N-fragment (147.9 (79.0–257.8) vs. 206.5 (99.3–440.8) pmol/mg Cr in unchanged vs. combination,
p
Diaphragm dysfunction is preventable and reversible. Therefore, it is important to maintain spontaneous breathing and avoid excessive ventilatory support during mechanical ventilation, which is called diaphragm protective ventilation [37]. Diaphragm protective ventilation can prevent diaphragm atrophy, compared with lung protective ventilation [52]. Furthermore, O’ Rourke et al. reported that percutaneous electrical phrenic nerve stimulation increased diaphragm thickness by 15.1% within 48 h [53]. Extracorporeal support is also considered to prevent diaphragm injury [54], and in a case report, the early initiation of extracorporeal support prevented diaphragm atrophy, with a relatively suppressed level of urinary titin N-fragment of 24.1–38.4 pmol/mg Cr [55].
2.3. Other Respiratory Muscle Atrophy
In addition to the diaphragm muscle, intercostal muscle atrophy is also observed in patients with mechanical ventilation [56], and is associated with prolonged mechanical ventilation and prolonged ICU stay [36]. Moreover, muscle atrophy occurs in other expiratory muscles, including the obliquus interna, obliquus externa, transversus abdominis, and rectus abdominis muscles [57]. In the case report of a mechanically ventilated patient, intercostal muscle biopsy showed the loss of myosin-containing thick filaments, with the possible detachment of titin [58]. Titin loss may be an important cause of other respiratory muscle dysfunctions, as well as that of the diaphragm. Jonkman et al. reported that breath-synchronized electrical stimulation increased the thickness of abdominal expiratory muscles (1.76 mm vs. −0.50 mm in intervention vs. control, respectively,
p = 0.02) [59]. Thus, titin loss may be reversible by active rehabilitation.