Hydrogen sulfide (H
2
S) is an endogenously produced gaseous signaling molecule and is critical for the regulation of cardiovascular homeostasis.
- heart
- cardiovascular disease
- heart failure
1. Introduction
Hydrogen sulfide (H2S) is an endogenously produced gaseous signaling molecule and is critical for the regulation of cardiovascular homeostasis [1][2][1,2]. It is mostly produced enzymatically in mammalian species by three enzymes in the cysteine biosynthesis pathway: cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST). New enzymes are emerging as important contributors to H2S production. Apart from these enzymes, H2S can also be produced from sulfur reservoirs (i.e., sulfane sulfur). Clinically, there is a negative association between heart failure, diabetes and H2S, as evidenced by the findings that lower circulating H2S levels are detected in plasma samples taken from diabetic and heart failure patients [3][4][5][3,4,5]. Therapeutic strategies aimed at increasing the levels of H2S are protective in models of acute myocardial ischemia-reperfusion injury and heart failure [6][7][8][9][6,7,8,9]. Numerous studies have shown the key role of H2S in maintaining cardiovascular homeostasis, but many of these studies rely on exogenous H2S donors. More studies that provide novel insight into endogenous H2S dynamics can help investigators understand the role of H2S in heart physiology.
2. Overview of Hydrogen Sulfide
2.1. The Molecule and Post-Translational Modification
Originally described as a toxicant, H2S has emerged as an essential gaseous molecule that is both clinically and physiologically relevant to the heart. While the chemistry of this molecule is reviewed elsewhere (see Filipovic et al. (2018)), in this section we will briefly revisit important properties [10][12].
H2S is a volatile, water-soluble molecule with the bond dissociation energy similar to thiols (90 kcal/mol vs 92 kcal/mol, respectively). This property indicates that H2S does not readily decompose, making it a good signaling molecule [11][13]. Although H2S is membrane diffusible with a diffusion capacity from 0.5–10 cm/s, as a reactive electrophile, intracellular factors may regulate its travel [12][14]. Autooxidation involving metal catalysts and oxygen that can extinguish the reactivity of the molecule and transition it into alternative oxidation states has been reported [11][13]. Importantly, H2S not only reacts with other oxidants, such as nitric oxide, but can modify oxidized thiols (e.g., sulfenic acid) to form PSSH [10][13][14][12,15,16]. The post-translational modification of proteins is the subject of active research to understand the molecular and physiological consequences.
For a protein to be S-sulfhydrated, the oxidation of H2S or the targeted thiol is required [12][14]. Though PSSH cannot shield proteins from oxidative damage, PSSH has a higher bond dissociation energy than other oxidative modifications, such as S-nitrosation (PSSH: 70 kcal/mol vs. 31–32 kcal/mol) [13][15]. This may allow PSSH to serve as a more stable signaling modification [15][16][17][17,18,19]. While significant advances in proteomics have identified several proteins to be dynamically S-sulfhydrated under various physiological conditions, more studies are needed to further understand the impact of PSSH and its regulation of cardiac metabolism and disease [18][19][20][21][20,21,22,23].
2.2. Mechanisms of Hydrogen Sulfide Production
An important node for H2S regulation is in its production. L-cysteine, generated from the cysteine biosynthesis pathway, is the fundament substrate for enzymatic H2S production. Three enzymes are primarily responsible for H2S generation: CBS, CSE, and 3-MST (Figure 1). Both CBS and CSE utilize cystathionine in a pyridoxal 5′ phosphate (PLP)-dependent reaction. On the other hand, in the cysteine catabolism pathway, L-cysteine is converted into 3-mercaptopyruvate by cysteine aminotransferase in a PLP-dependent manner and is then metabolized by 3-MST into pyruvate and H2S. Apart from these enzymes, others are thought to participate in transsulfuration and facilitate H2S production. Together, these enzymes form key nodes to produce and regulate endogenous H2S production. While the enzymology has been extensively studied, few studies have shown the role of these enzymes (except CSE) in cardiac biology.
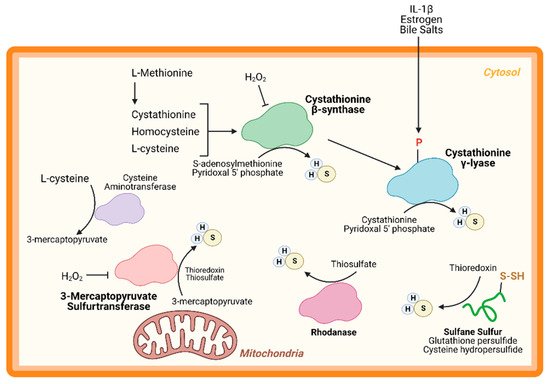
Figure 1. Hydrogen sulfide (H2S) Production. H2S is predominately produced by three enzymes—cystathionine-β-synthase, cystathionine-γ-lyase, and 3-mercaptopyruvate sulfurtransferase—and can be generated from other processes, such as sulfide quinone oxidoreductase and sulfane sulfur reservoirs (e.g., glutathione/cysteine (hydro)persulfides). H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation.
CBS is a key enzyme in the reverse transsulfuration pathway that regulates the flux of sulfur from the methionine cycle to the cysteine and glutathione biosynthesis pathways. In humans, CBS is a PLP-dependent tetramer that condenses cysteine and homocysteine into cystathionine with a heme co-factor to release H2S (Figure 1). CBS can also condense cysteines to produce H2S and lanthionine. Its activity is limited by the concentration of cysteines and is regulated by S-adenosylmethionine [22][23][24,25]. Oxidative stress conditions can lead to a truncated, S-adenosylmethionine-insensitive form of CBS that has increased activity, yet other studies suggest that the oxidation of its N-terminal heme group or CXXC oxidoreductase motif may inhibit the enzyme [24][25][26][26,27,28]. More studies are needed to understand the role of CBS in cardiac biology and disease. While few, if any, studies have proven a cardioprotective role for CBS, mutations in this protein can lead to hyperhomocysteinemia and an increased risk for cardiovascular disease along with accelerated atherosclerosis [27][29].
CSE is the predominant H2S-generating enzyme in the cardiovascular system and its ability to remediate cardiac injury is under active investigation [28][30]. Like CBS, human CSE exists in a PLP-bound, tetrameric state that, apart from H2S, yields α-ketobutyrate, pyruvate, and ammonia (Figure 1) [29][31]. CSE is also sensitive to homocysteine levels [26][28]. CSE is a key player in protection from various diseases, including heart disease. Several studies have demonstrated that CSE inhibition or genetic deficiency increased infarct size and enhanced transaortic constriction (TAC)-induced heart failure severity [30][31][32,33]. Conversely, cardiac-specific CSE overexpression blunted TAC-induced heart failure progression [32][34]. Also, CSE and PSSH was shown to decrease in the aged heart [13][15]. Taken together, these studies demonstrate that CSE is an integral component of both cardiovascular disease and disease risk.
Recently, CSE was found to be phosphorylated during atherosclerosis, leading to a decrease in H2S production [33][35]. Conversely, 17β-estradiol, the active form of estrogen, stimulates H2S production by CSE phosphorylation (Human Ser56) in a cyclic guanosine monophosphate/protein kinase G-dependent mechanism [34][36]. Also, bile salts stimulate CSE phosphorylation in endothelial cells [35][37]. This finding shows that H2S produced by CSE can be regulated by post-translational modifications (Figure 1). CSE has been shown to translocate to the mitochondria during endoplasmic reticulum stress and helps to maintain adenosine triphosphate (ATP) production. Furthermore, this translocation is Tom20 dependent and may help to regulate cystathionine levels in the mitochondria [36][38].
3-MST is a homodimeric, PLP-independent enzyme that receives 3-mercaptopyrvuate from cysteines metabolized by a PLP-mediated reaction via cysteine aminotransferase (CAT/GOT). Also, in the brain and kidneys, in a PLP-independent manner, D-cysteine can contribute to 3-mercaptopyruvate production via D-amino acid oxidase [37][39]. Upon binding, 3-MST converts 3-mercaptopyruvate into sulfane sulfur, then either thioredoxin or a thiol-based reductant liberates H2S and pyruvate (Figure 1). While CBS and CSE are predominately cytosolic, 3-MST is reportedly found in both the cytosol and mitochondria, suggesting that 3-MST may be a key protein that regulates mitochondrial protein PSSH.
While phosphorylation, or other post-translational modifications have not yet been reported, or investigated, 3-MST is reportedly regulated in two ways: oxidative stress and dimerization (Figure 1). Oxidative stress from hydrogen peroxide inhibited 3-MST activity in cultured hepatoma cells, suggesting that disease which stimulate excess oxidation may suppress H2S production from 3-MST [38][40]. Furthermore, redox-sensitive cysteines within 3-MST can govern its dimerization [39][40][41][41,42,43]. Oxidation of 3-MST can induce dimerization rendering the enzyme inactive, however—evident from treatment with reducing agents and shielding cysteines from oxidation with an alkylating agent—3-MST monomerization maintains H2S production [39][41][41,43].
While CBS, CSE, and 3-MST are the most widely studied enzymes, others have also been identified. Rhodanese, or thiosulfurtransferase, participates in a transsulfuration reaction by catalyzing the transfer of sulfur from thiosulfate to thiol or cyanide to form persulfides or thiocyanate, respectively (Figure 1) [42][44]. This protein closely resembles 3-MST and may be functionally cooperative [43][44][45,46]. Interestingly, 3-MST knockout mice are reported to have a 3-fold increase in rhodanese expression that may be an adaptive response to the loss of 3-MST, however, the consequences of excessive rhodanese in these mice are not yet understood [45][47]. Conversely, mutation of key residues in rhodanese decreased its activity and increased 3-MST activity [46][47][48,49]. While the role of 3-MST in cardiac homeostasis and disease has been recently reported, to our knowledge, no study has yet to investigate the importance of rhodanese in cardiac biology.
Sulfane sulfurs are H2S reservoirs bound to proteins (e.g., PSSH) or other molecules and are important sources of H2S. Cysteine hydropersulfides and glutathione persulfides (GSSH) are produced endogenously and display properties for maintaining intracellular homeostasis (Figure 2) [48][50]. PSSH are reduced, or de-sulfhydrated, by thioredoxin to release H2S [21][49][23,51]. Sulfate and sulfite are also important H2S reservoirs. GSSH is reduced to sulfite by persulfide dioxygenase, then further oxidized to by sulfide oxidase (SO). Through sulfite production, at least with SO deficiency, GOT can also produce H2S by catalyzing the deamination of cysteine sulfinic acid, the first product of oxidative cysteine metabolism [50][52]. While there have been advances in H2S biology, more studies are needed to fully comprehend the complexity of this redox molecule, especially regarding its role in cardiac metabolism.
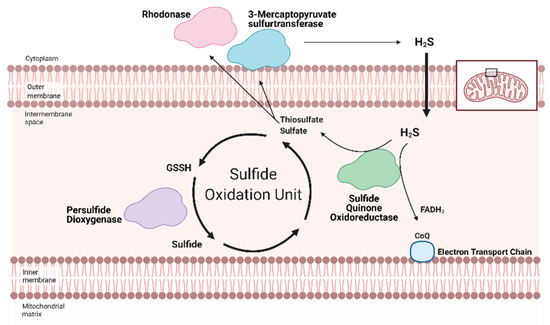
Figure 2. Hydrogen Sulfide (H2S) catabolized by the Sulfide Oxidation Unit. Free H2S can be removed from the cell either by storage in sulfane sulfur pools or the Sulfide Oxidation Unit. H2S catabolism is linked to mitochondrial respiration via sulfide quinone oxidoreductase (SQR). Nicotinamide adenosine dinucleotide + hydrogen (NADH) and flavin adenosine dinucleotide + 2 hydrogen (FADH2) reduce Complex I and II, respectively. The electrons travel across the Electron Transport Chain, through Complex III and cytochrome c (Cyt c) to ultimately reduce oxygen in Complex IV. This flow generates a hydrogen gradient that drives adenosine triphosphate (ATP) synthase and allows for ATP generation from adenosine dinucleotide (ADP). SQR, a key part of the Sulfide Oxidation Unit, extracts electrons from H2S and reduces co-enzyme Q (CoQ), which transfers the electrons to Complex III, contributing to ATP production. As a byproduct, SQR also produces thiosulfate and sulfate—the former can be converted back into H2S by 3-mercaptopyruvate sulfurtransferase. As part of the Sulfide Oxidation Unit, thiosulfate and sulfate react with glutathione to form glutathione persulfide (GSSH), which is then converted into sulfide and recycled back into thiosulfate. Adapted from “Electron Transport Chain”, by BioRender.com (accessed on 24 February 2021). Retrieved from https://app.biorender.com/biorender-templates(accessed on 24 February 2021).
3. Hydrogen Sulfide in Heart Metabolism and Cardiac Disease
3.1. Overview
The heart relies on fatty acids to generate energy, while other organs utilize glucose as their primary energy substrate [51][52][53,54]. The energy demand of the heart requires the use of fatty acids, because this substrate yield more ATP molecules than glycolysis [53][55]. Long-chain fatty acids are transported into cells by fatty acid translocase, CD36 [54][56]. Once inside the cell, fatty acid oxidation (FAO) is initiated with the transport of long-chain fatty acids that are modified by long-chain acyl-co-enzyme A (CoA) synthetase (LACS) and carnitine palmotyltransferase 1 (CPT1). LACS cleaves an acyl-CoA molecule and delivers it to CPT1 [55][57].
CPT1 is a rate-limiting enzyme of FAO that controls the entry of acyl-CoA into the mitochondria. In cardiomyocytes, CPT1b is the predominant isoform, accounting for 98% of CPT1 activity, while in other organs CPT1a (e.g., liver) and CPT1c (neurons) are more active [56][57][58][59][60][58,59,60,61,62]. CPT1 converts acyl-CoA into acylcarnitine and transports the product across the outer mitochondrial membrane. Then, carnitine acylcarnitine translocase (CACT) allows acylcarnitine to traverse the inner mitochondrial membrane and react with CPT2. With CPT2, acylcarnitine is converted back into acyl-CoA and enters the tricarboxylic acid (TCA) cycle [55][57].
Briefly, the TCA cycle is amongst the most well-studied and most important metabolic cycles in biology. The churn begins with the condensation of acyl-CoA into citrate, then the cycle carries citrate to oxaloacetate. Along the way, the TCA cycle produces two essential reducing equivalents—nicotinamide adenosine dinucleotide + hydrogen (NADH) and flavin adenosine dinucleotide + 2 hydrogen (FADH2)—along with other metabolites that participate in other metabolic systems. Both NADH and FADH2 feed electrons into the Electron Transport Chain that ultimately reduces oxygen and drives the migration of hydrogen over the mitochondrial membrane, power ATP synthase, which generates ATP (Figure 2). From one C18 fatty acid, FAO can yield 120 ATP molecules versus the same amount of ATP from three glucose molecules [53][55].
The heart beats ceaselessly throughout a human’s, or animal’s, life and generates pressures needed to overcome vascular resistance. This process requires enough energy to sustain homeostasis for both the cardiomyocytes and the other cells in the body. FAO gives the heart the energy it needs to function, however disease can the shift the metabolic substrate used and often forces the heart to utilize glycolysis instead of FAO [61][63]. H2S may be a way to preserve cardiac metabolism and remediate cardiovascular disease.
3.2. Hydrogen Sulfide Regulation Integrated with Mitochondrial Respiration
Redox homeostasis requires both the production of reactive molecules that can alter cellular function and a means of controlling that production. H2S regulation is intimately linked with metabolism and mitochondrial respiration (as reviewed by Paul et al. (2021)) [62][64]. Although H2S can suppress metabolism at high doses, recent studies show that low dose H2S is a metabolic stimulator, in part by providing electrons for mitochondrial respiration [63][64][65][66][65,66,67,68]. At low concentrations, H2S can serve as an inorganic electron donor—as it does in microorganisms—to Coenzyme Q with the help of sulfide quinone oxidoreductase (SQR) [62][67][64,69].
Along with persulfide dioxygenase and rhodanese, SQR is a key component of the Sulfide Oxidation Unit, which is responsible for the catabolism of free H2S (Figure 2) [67][68][69,70]. H2S is oxidized by SQR to yield two electrons that are transferred to FAD, then Coenzyme Q, which reduces Complex III [69][71]. This reaction also produces thiosulfate or sulfate that reacts with reduced glutathione to form GSSH. This new persulfide on glutathione is then oxidized to sulfide by persulfide dioxygenase. Sulfide can be further metabolized by either sulfide oxidase to sulfate or rhodanese to a thiosulfate. Thiosulfate can be catabolized back into H2S by 3-MST or rhodanese [70][71][72,73]. The cyclical nature of these reactions not only controls the toxic accumulation of H2S but may help to maintain a physiologic pool of sulfur that can be used for important redox reactions and other essential molecular processes.
Various cardiac diseases are linked to decreased H2S production and availability. Under active investigation are mechanisms that regulate H2S production and the ways they are impaired during disease. Below, we focus on two cardiac diseases well understood to suppress H2S production and availability, and their linked to H2S-mediated metabolic regulation: diabetic cardiomyopathy and heart failure.
3.3. Diabetic Cardiomyopathy and Hydrogen Sulfide
The prevalence of obesity, insulin resistance, diabetes, and dyslipidemia are increasing worldwide. People who have these disorders, collectively called metabolic syndrome, are twice as likely to develop heart failure and have worse prognoses after cardiovascular disease development [72][74]. In the U.S., obesity has become a major risk factor for metabolic syndrome development. From 2013–2019, 34% of overall children (2–19 years old) and 38% of adults in the U.S. were obese. Notably, the highest prevalence was amongst Hispanics [73][75]. Studies implicate “Western” diets, or high-fat diets (HFD), as a key driver of obesity [74][76]. Most patients with metabolic syndrome have hypertriglyceridemia and increased plasma levels of fatty acids. These lipids are taken into the heart, where due to an overload in storage capacity and utilization, accumulate and become lipotoxic to the cardiomyocytes. This induces a non-ischemic form of cardiomyopathy termed lipotoxic or diabetic cardiomyopathy (DCM) that can progress to heart failure [72][75][74,77].
Metabolic syndrome is strongly associated with a significant decrease in H2S availability and protein PSSH [3][4][3,4]. This syndrome leads to DCM due to the accumulation of lipids that become toxic. Oxidative stress and inflammation merge with toxic lipid accumulation to form more oxidants that overwhelm the metabolic system of the heart and kill the irreplaceable cardiomyocytes [72][74]. Cell death then leads to pathologic hypertrophy and can decompensate into heart failure [76][77][78,79]. This non-ischemic form of cardiomyopathy is linked to dysregulated metabolism and suppressed H2S production.
Clinically, there is a negative association between diabetes and H2S, evident from the findings that lower circulating H2S levels are detected in plasma samples taken from diabetic patients [3][4][3,4]. Preclinical models, specifically HFD-fed mice or fatty acid-treated cell models, have replicated these findings. Recent animal work showed that lost H2S bioavailability (e.g., lowered CSE expression, reduced protein PSSH, and impaired H2S production) contributes to the etiology of metabolic syndrome [78][79][80][81][82][83][84][80,81,82,83,84,85,86]. Additionally, cell models treated with toxic lipids, such as palmitate, reported impaired H2S production and reduced protein PSSH [85][87]. Many studies have shown that exogenous H2S can protect against excessive heart injury, including DCM [86][87][88][88,89,90]. While H2S donors are a promising form of therapy, stimulating endogenous H2S production is an innovative strategy that may improve target specificity.
Few studies have studied the underlying mechanisms that govern H2S-mediated regulation of metabolism; however, this is an active area of research. H2S can not only donate to the mitochondrial respiration, but it can also modify ATP production. Under stress conditions, mitochondrial CSE translocation was shown to increase mitochondrial H2S and enhanced ATP production in smooth muscle cells [36][38]. ATP5A1, a subunit of ATP synthase, is also known to be S-sulfhydrated in a CSE-dependent manner [89][91]. In streptozotocin-induced, diabetic rats, 3-mercaptopyruvate—the substrate for 3-MST—was injected intraperitonially and elevated circulating H2S, along with stimulating oxygen consumption from liver mitochondria [90][92]. These studies suggest that elevating mitochondrial H2S may be a protective adaptation to regulate bioenergetics. Apart from mitochondrial respiration, our group has shown that H2S can stimulate carbohydrate and lipid metabolism. Through the activation of adenosine monophosphate kinase (AMPK) by S-sulfhydrating and inhibiting scaffolding protein phosphatase 2A (PP2A), the heart upregulated metabolic genes (Figure 1) [91][93]. Recently, Bithi et al. (2019) described a cardiac and endothelial S-sulfhydryl proteome [18][92][20,94]. Several proteins involved in metabolism were identified, yet the impact of PSSH on enzymatic activity and cardiac metabolism remains to be understood.
A way that H2S can regulate metabolism is by modulating transcription factors activation, specifically those responsible for the upregulation of metabolic genes (Figure 3). Peroxisome proliferator-activated receptors (PPAR) are major transcription factors that control the expression of FAO and uptake genes in all organs [93][94][95,96]. Both PPAR-δ and PPAR-γ are modified and activated by H2S, inducing lipid metabolism [95][96][97,98]. Our group has shown that peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α, a key co-regulator of PPAR signaling is induced by H2S treatment through the stimulation of AMPK [91][93]. Hypoxia-inducible factor (HIF)-1α is an essential transcription factor that stimulates the expression of glycolytic genes in response to hypoxia or oxidation [97][99]. Under normoxic conditions, CBS-dependent H2S inhibits prolyl hydroxylase 2, which helps target HIF-1α for degradation, thus allowing HIF-1α to accumulate and induce gene expression [98][99][100,101]. These important proteins regulate the expression of metabolic genes demonstrating that H2S can even regulate metabolism at the transcriptional level.
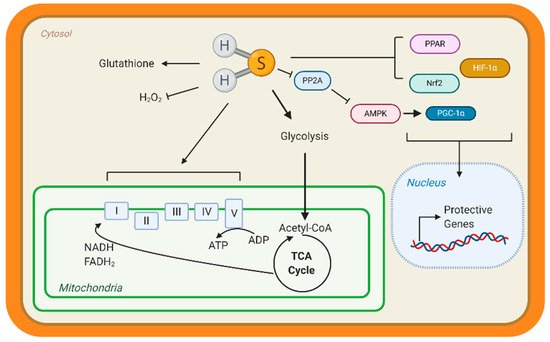
Figure 3. Hydrogen sulfide (H2S) Metabolic Targets in the Heart. Few studies have identified metabolic targets of H2S, but those that have been identified are key for cardiac metabolism. H2S can modify free cysteines on enzymes, a reaction known as S-sulfhydration, to alter metabolic pathways, such as glycolysis, and induce up-regulation of metabolic genes via transcription factor activation. Peroxisome proliferator-activated receptors (PPAR), hypoxia-inducible factor 1α (HIF-1α), and nuclear factor E2-related factor 2 (Nrf2) are transcription factors known to be S-sulfhydrated and regulate heart metabolism. H2S also modified the complexes of the Electron Transport Chain and can stimulate mitochondrial respiration. H2S is considered an antioxidant molecule because it can react with glutathione and hydrogen peroxide (H2O2). Finally, our group found that S-sulfhydration represses protein phosphatase 2A (PP2A) activity, allowing for the activation of adenosine monophosphate kinase (AMPK) and phosphorylation of the transcription factor peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) to induce gene expression.
Few studies have focused on the impact that dysregulated H2S can have on cardiac metabolism and disease. The field needs more innovative experiments and approaches that capture the dynamics of H2S production and signaling to understand the beneficial nature of this gaseous molecule. Harnessing this endogenous donor pool may help reduce the burden of cardiovascular disease by adding specificity to the existing body of exogenous H2S donors. A way to study these dynamics may be to study the underlying molecular mechanism in the development of metabolic syndrome and the interventions that reduce cardiovascular disease risk from metabolic disorders.
3.4. Heart Failure
According to the annual American Heart Association statistics, about 6.2 million Americans over the age of 20 were affected with heart failure between 2013–2016, and that number is expected to rise with an aging population. Heart failure is projected to increase by 46% between 2012–2030 [100][102]. Racial, ethnic, and gender disparities exist with heart failure. In the Health, Aging and Body Composition Study, the risk of developing heart failure the elderly black population—attributed to modifiable risk factors, such as smoking and blood pressure—was about 68% compared to 49% for the elderly white population [101][103]. The Hispanic population, which also has a high cardiometabolic risk—as previously described—also has a high risk of developing heart failure [102][103][104,105]. While women tend to have more co-morbidities, such as diabetes and high blood pressure, heart failure mortality prognosis is better for women compared to men [104][105][106,107]. Reducing the burden of heart failure in the aging population is imperative and molecular insights may help to develop preventative interventions that can lower heart failure risk.
Heart failure—a consequence of DCM—is characterized as a significant loss of cardiac output due to a substantial alteration in heart structure [106][108]. The etiology of heart failure can come from various sources, mostly from events that injure the heart and results in the death of irreplaceable cardiomyocytes. As previously described, DCM is a non-ischemic form of cardiomyopathy and over time results in heart failure, however ischemic injury also reduces cardiac function. Myocardial infarctions—cardiac muscle death usually resulting from an occlusion of the coronary arteries which feed the heart—and subsequent reperfusion is an example of ischemic injury that can lead to heart failure [107][109]. The cardiomyocytes of the injured heart swell and undergoes pathological hypertrophy to compensate for a loss in cardiac output, however this adaptation is unsustainable [108][109][110,111]. Over time, the muscle degrades, the heart scars, and decompensates into heart failure [76][77][78,79].
Heart failure manifests as symptoms of dyspnea (labored breathing) and pulmonary congestion (i.e., edema) among others, resulting from significant physiological alterations [106][108]. A consequence of reduced cardiac output is a drop in blood pressure. This stimulates the brain to increase β-adrenergic signaling by releasing more norepinephrine and epinephrine from the adrenal glands. Meanwhile, the kidneys initiate the renin-angiotensin system to induce vasoconstriction, which can lead to kidney disease [110][111][112,113]. Both adaptations increase blood pressure to healthy levels, attempting to elevate organ perfusion and compensating for the loss of cardiac output. However, the heart is not as it once was; this “high” blood pressure further strains the weakened heart, killing more cardiomyocytes [108][112][110,114].
The role of endogenous H2S production and regulation is not completely understood in heart failure. Heart failure can be categorized into heart failure with reduced or preserved ejection fraction (HFrPF or HFpPF, respectively) [113][114][115,116]. While reports are emerging that focus on HFpPF, a large body of literature has described the molecular mechanisms involved in HFrPF. In severe, end-stage heart failure patients, there is a marked reduction in circulation H2S that is reproduced in pressure-induced heart failure animal models, such as TAC [5]. Furthermore, several studies have shown that pharmacological donors of H2S, such as sodium sulfide, SG-1003, among others, preserve cardiac function in pressure-induced and ischemia-induced heart failure animal models [9][115][116][117][9,117,118,119]. Heart failure in CSE deficient mice was more severe compared to WT controls [32][34]. These studies strongly implicate H2S as a key mechanism of heart failure.
In animal models, heart failure can be induced by pressure overload with TAC—banding of the aorta to simulate excess arterial pressure. Also, surgical occlusion of a coronary artery—cardiac vessels that feed the heart—can cause ischemia. Our group showed that sodium sulfide protected hearts from ischemic injury in a Nuclear factor E2-related factor 2 (Nrf2)-dependent manner (Figure 3). H2S-mediated activation of this transcription factor, which is responsible for upregulating antioxidant responses, sustained proteosome activity, thus eliminating dysfunctional proteins and preserving cellular homeostasis [117][118][119,120]. Moreover, H2S stimulated pro-angiogenic factors, such as nitric oxide bioavailability and vascular endothelial growth factors, increasing cardiac vascular density [115][117]. In culture cardiomyocytes, H2S attenuated hypertrophy and stimulated glycolysis by upregulating glucose transporter 4, along with increasing pyruvate kinase and succinate dehydrogenase activity [119][121]. Also, age-dependent cardioprotection against ischemia-reperfusion injury was reported in young 3-MST knock out mice, while 18-month-old mice showed hypertension and cardiac hypertrophy [120][122]. Taken together, H2S donors preserve cardiac structure and function, however endogenous H2S—evident from CSE deficient mice—also contributes to protection.
While H2S donors remain a vital strategy to efficently deliver this potent and protective molecules, we propose that exploiting the endogenous, cardiac H2S system can also be an integral and specific way of remediating heart failure and other cardiovascular diseases. Historically, the benefits of H2S of heart disease were demonstrated by donors, because—until recently—few stimulators of endogenous H2S were known. These H2S donors are non-specific because they release H2S throughout the body or an organ. H2S-producing enzymes are naturally positioned throughout and likely target specific, redox-sensitive proteins initiate a cardiac stress response, thus confer protection against disease. Targeting these H2S enzymes and modifying their activity—without genetic manipulation, such as removal or overexpression—may be a way to develop targeted therapeutics. However, few studies have investigated endogenous H2S regulation during disease. Of the few reports, cultured hepatocytes supplemented with 3-mercaptopyruvate—the substrate for 3-MST—stimulated 3-MST- and SQR-dependent H2S production. This treatment increased mitochondrial bioenergetics, yet the experiments were not set in the context of disease [121][123]. The role of endogenous H2S in the heart remains to be fully understood, yet there is clearly a role for H2S enzymes, such as 3-MST, in maintaining cardiac homeostasis during disease. Physiological stimulators of cardiac H2S are emerging, and these stimulators are well-known interventional strategies, which reduce the risk of cardiovascular disease.