TIGIT is a transmembrane glycoprotein comprising one immunoglobulin variable (IgV) domain, a type I transmembrane domain, and a cytoplasmic tail with an immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoglobulin tyrosine tail (ITT)-like motif.
- cancer immunotherapy
- immune checkpoint blockade
- TIGIT
- PVR
1. TIGIT Structure and Its Ligands
TIGIT is a transmembrane glycoprotein comprising one immunoglobulin variable (IgV) domain, a type I transmembrane domain, and a cytoplasmic tail with an immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoglobulin tyrosine tail (ITT)-like motif [1][2][3][17,18,19]. The cytoplasmic tail of TIGIT initiates an inhibitory signaling cascade. Previous studies have reported that ITT-like motif (Tyrosine, Y225) mediates a major inhibitory signal in humans, whereas mouse TIGIT inhibitory signal can be triggered by either the ITIM (Y277) or the ITT-like motif residue (Y233) [4][20]. Upon binding to its ligand, the cytoplasmic tail of TIGIT is phosphorylated and binds to cytosolic adaptor growth factor receptor-bound protein 2 (Grb2), recruiting Src homology 2 (SH2)-containing inositol phosphate-1 (SHIP-1). SHIP-1 inhibits phosphoinositide 3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) signaling cascades [5][21]. Moreover, phosphorylated TIGIT associates with beta-arrestin 2 and recruits SHIP-1, which further suppresses the auto-ubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF-6) to inhibit nuclear factor kappa B (NF-κB) activation [5][6][21,22].
TIGIT has multiple ligands, including PVR (Necl-5 or CD155), nectin-2 (CD112), nectin-3 (CD113), and nectin-4 (PVRL4) [7][8][13,23]. Nectin and Necl proteins are cell-surface glycoproteins that belong to the immunoglobulin superfamily. Nectin family comprises four members (nectin-1–4), and the Necl family consists of five members (Necl-1–5). They have three Ig ectodomains, which form homodimeric or heterodimeric complexes in the membrane [9][24]. The IgV domain of TIGIT exhibits sequence homology with PVR, nectin-1, nectin-2, nectin-3, and nectin-4 [1][17]. TIGIT binds to PVR with high affinity and nectin-2 and -3 with low affinity. Recently, nectin-4 has been reported to bind to TIGIT with an affinity similar to that of TIGIT and PVR binding [10][25]. PVR plays immunoregulatory roles by interacting with TIGIT, CD226, and CD96 [11][12][13][26,27,28]. PVR has a greater affinity for TIGIT than either CD226 or CD96, implying a dominant role of TIGIT inhibitory signaling over activation signals. Furthermore, PVR expression is commonly upregulated in several types of cancer and tumor-associated myeloid cells [14][15][29,30]. Elevated PVR expression has been associated with an unfavorable prognosis across various solid cancer types [16][17][31,32]. Nectin-2 interacts with TIGIT, CD226, and CD112R; however, both TIGIT and CD226 have much weaker binding affinity to nectin-2 than PVR. Similar to PVR, the TIGIT–nectin-2 interaction could transduce an inhibitory signal, but the CD226–nectin-2 interaction triggers immune cell activation. A recent study has demonstrated that the inhibitory effect of nectin-2 is mediated by CD112R and not TIGIT [18][14].
2. Role of TIGIT in Immune Cell Regulation
TIGIT is expressed on most NK and multiple T cell subsets, including memory and activated T cells, regulatory T cells (Treg), and follicular T helper cells (TFH) [1][3][4][17,19,20]. Upon activation with its ligands, TIGIT expression is upregulated in both T and NK cells, where TIGIT inhibits cytotoxic activity. TIGIT-deficient mice do not develop spontaneous autoimmunity; however, they exacerbate experimental autoimmune encephalitis when immunized with myelin oligodendrocyte glycoprotein, indicating a suppressive role of TIGIT [12][27]. In preclinical mouse tumor models, TIGIT deficiency delays the subcutaneous growth of both B16F10 and MC38 cells and lung metastasis of B16 cells [19][20][33,34]. Moreover, TIGIT-deficient mice show increased survival when challenged with VK*MYC myeloma cell lines [21][35]; however, a recent study revealed that TIGIT-deficient mice did not reject the implanted B16F10 and MC38 more efficiently compared with wild-type (WT) mice [22][36]. Moreover, in B16F10, RM-1, and E0771 cell lung metastasis models, the beneficial effect of TIGIT deficiency on tumor metastasis was not observed [23][24][37,38]. These discrepancies might be results of different experimental setups and/or mouse housing conditions [25][39]. Further studies with immune cell-type-specific TIGIT-deficient mouse models would be helpful to clarify the suppressive role of TIGIT in vivo [20][34].
Several mechanisms may explain TIGIT-mediated inhibition of T and NK cell activities. First, as aforementioned, TIGIT delivers an inhibitory signal resulting from ITIM and/or ITT motifs within its cytoplasmic domain. Agonistic anti-TIGIT antibodies inhibit human and mouse T cell proliferation and cytokine production without antigen presenting cells (APC) by suppressing T cell receptor/CD28-activating signaling [12][26][27,40]; however, TIGIT engagement increases the expression of receptors for T cell maintenance (e.g., interleukin [IL]-2R, IL-7R, and IL-15R) and anti-apoptotic molecules (e.g., Bcl-xL) [12][27], implying that TIGIT signaling could mediate the survival of Tex cells. Additionally, TIGIT signaling also inhibits cytotoxicity, degranulation, and cytokine secretion of NK cells [3][27][19,41]. Moreover, TIGIT disrupts CD226 co-stimulation. TIGIT has higher affinity for the same set of ligands (PVR and CD112) than CD226. Thus, TIGIT outcompetes CD226 for binding to its ligands [1][17]. Knockdown of TIGIT in human CD4+T cells induces T-bet-mediated interferon (IFN)-γ production, which can be overcome by blocking CD226-CD155 signaling [26][40]. Additionally, TIGIT hinders CD226 signaling through the physical prevention of CD226 homodimerization [28][42]. A recent study by Jin et al. has demonstrated that TIGIT directly affects the intracellular regulation of CD226 activation. By using an antibody specifically recognizing the phosphorylated form of CD226 (phospho-Y322), they have shown that CD226 phosphorylation at Y322 is reduced in TIGIT WT-expressing Jurkat cells upon PVR engagement but not in the cells expressing TIGIT mutant (Y225A/Y231A) [29][43]. In addition, TIGIT has been known to suppress T cell function in a cell-extrinsic manner. Following TIGIT ligation, PVR signaling leads to increased production of IL-10 and diminished production of IL-12p40 in human dendritic cells (DCs), which further downregulates T cell activation [1][17]. In accordance with this result, TIGIT ligation inhibits macrophage activation and leads to increased M2 macrophage polarization through PVR [30][44].
The role of TIGIT has been implicated in modulating Treg cell responses. [31][32][45,46]. TIGIT expression is observed in a subset of natural Treg cells in both mice and human. TIGIT+Treg cells express higher levels of Treg signature genes, including Foxp3, CD25, and CTLA-4, compared with TIGIT−Treg cells. TIGIT expression is strongly correlated with the suppressive capacity and the lineage stability of human Treg cells [31][32][33][45,46,47]. Furthermore, TIGIT engagement leads to the induction of IL-10 and fibrinogen-like protein 2, which selectively suppress T helper type 1 (Th1) and Th17 responses [31][45].
3. Targeting TIGIT for Cancer Immunotherapy
3.1. TIGIT as a Potential Prognostic Marker for Cancer
Accumulating data from the immune monitoring of cancer patients have revealed that TIGIT expression is elevated in T and NK cells, and it often appears to be associated with advanced disease status and poor clinical outcomes [20][21][34][35][36][37][38][39][40][41][42][43][44][34,35,48,49,50,51,52,53,54,55,56,57,58]. In follicular lymphoma (FL) patients, TIGIT is highly expressed on intratumoral Treg and late-stage memory CD8+T cells, and increased numbers of TIGIT-expressing tumor infiltrating T cells reveal a correlation with poor survival rate [34][48]. Multidimensional flow cytometric analysis of intratumoral T cells obtained from FL patients before and after anti-PD-1 therapy has revealed that TIGIT+ Tex cells majorly respond to this therapy. It has been observed that TIGIT+ exhausted T cell populations are downregulated and TIGIT+ effector cells are upregulated by anti-PD-1 therapy [34][48]. Increase in the proportion of highly suppressive tumor-infiltrating Treg cells following TIGIT expression is associated with poor clinical outcomes in patients with hepatocellular carcinoma (HCC) and metastatic melanoma [33][43][47,57]. Moreover, upregulation of TIGIT indicates unfavorable disease status. High-risk patients with myelodysplastic syndrome (MDS) express higher levels of TIGIT and PD-1 in peripheral blood T and NK cells than low-risk patients [44][58]. High TIGIT expression renders CD4+T, CD8+T, and NK cells hypo-responsive to stimulation in high-risk MDS patients. Several studies have reported that TIGIT upregulation after treatment is correlated with recurrence. In patients with high-grade serous carcinoma, NanoString analysis of tumor tissues has indicated that recurrent tumors acquire a more inflamed phenotype with increased expression of TIGIT, CTLA4, Lag-3, and Tim-3 compared to primary tumors [45][59]. The proportion of TIGIT+CD8+T cells is increased in peripheral blood collected from acute myeloid leukemia (AML) patients, and it becomes more evident in patients with primary refractory disease and leukemia relapse post-allogeneic stem-cell transplantation [35][49]. Furthermore, TIGIT and/or PD-1 expression in CD8+T cells is increased in patients with gastric cancer relapse after treatment with SOX (S-1 and oxaliplatin) regimen, whereas no notable increase in the proportion of TIGIT+ and/or PD-1+CD8+T cells was found in relapse-free patients [46][60]. The compensatory increase in TIGIT expression post-treatment has also been observed in high-grade neuroendocrine neoplasms upon anti-PD-1 therapy [47][61].
3.2. TIGIT Blockade in Anti-Tumor Immunity
Based on the mechanism underlying TIGIT-mediated regulation of anti-tumor immune responses, efforts have been made to enhance T or NK cell activity by blocking TIGIT binding to its ligands, PVR and nectin-2, with monoclonal antibodies (mAbs) for therapeutic interventions. Several preclinical mouse models have been used to assess the anti-tumor efficacy of anti-TIGIT blocking mAbs. In CT26 colon carcinoma, EMT6 breast carcinoma, MC38 colon carcinoma, and GL261 glioblastoma models, treatment with anti-TIGIT-blocking mAbs combined with anti-PD-1 or PD-L1-blocking mAbs leads to nearly complete remission of tumor growth, whereas the treatment of anti-TIGIT mAbs as a single agent presents limited efficacy [28][48][49][42,62,63]. CD8+T cell depletion using anti-CD8α-depleting mAbs in CT26-bearing mice has revealed that the synergistic effect of dual blockade of TIGIT and PD-1 is mainly driven by the promotion of CD8+T cell responses. A triple combination of anti-TIGIT mAbs, anti-PD-L1 mAbs, and radiotherapy elicits almost complete remission of tumor growth in CT26-bearing mice [50][64].
Sufficient tumor regression by treatment with anti-TIGIT mAbs alone has been reported in different mouse tumor models. In multiple myeloma (MM) mouse tumor model, TIGIT blockade leads to reduced tumor growth and increased survival compared with mice receiving control IgG or anti-PD-1 mAbs [21][35]. Moreover, TIGIT blockade presents anti-tumor efficacy in Tgfbr1/Pten2 conditional knock-out (KO) mouse model that spontaneously develops head and neck squamous cell carcinoma (HNSCC) upon tamoxifen injection [41][51][55,65]. Both studies suggest that TIGIT is highly expressed on CD8+T and Treg cells in MM or HNSCC TILs and that anti-TIGIT mAbs reverse TIGIT-mediated suppression of CD8+T cell effector functions; however, the potent anti-tumor effect of anti-TIGIT mAbs as a single agent may not be fully guaranteed simply by the increased expression of TIGIT in TILs, since high TIGIT expression is also observed in CD8+ TILs in CT26-bearing mice that are not responsive to TIGIT blockade [28][42]. A recent study by Chiu et al. provided additional insights into the mechanism through which TIGIT blockade mitigates tumor immune evasion and resistance to PD-1 blockade [52][66]. They found that anti-PD-1 mAb treatment induced the upregulation of TIGIT in CD8+ TILs in Trp53 KO/C-MycOE mice, which is a highly aggressive HCC model; however, the compensatory expression of TIGIT upon PD-1 blockade was not observed in Hepa1-6-bearing mice that are known to be an anti-PD-1-sensitive orthotopic HCC model. PVRL1, which does not directly bind to TIGIT, contributed to TIGIT-mediated suppression of CD8+T cells by stabilizing PVR in HCC cells, and PVRL1 deficiency rendered HCC to be more sensitive to anti-PD-1 mAb treatment. In accordance with this finding, a differential level of the ligand expression, such as PVR and PD-L1, or an increase in the binding affinity of TIGIT to PVR under an acidic tumor microenvironment has been recently identified to contribute toward the sensitivity of tumor cells to TIGIT blockade [53][54][67,68].
Although TIGIT blockade is known to mainly act on CD8+T and Treg cells, NK cell dependent efficacy of anti-TIGIT mAbs is also suggested. A recent study by Zhang et al. reported that treatment with anti-TIGIT mAbs 3 days after tumor cell implantation prevented tumor-infiltrating NK cell exhaustion in CT26 or methylcholanthrene (MCA)-induced fibrocarcinoma-bearing mice, which resulted in the enhancement of CD8+T cell responses and tumor rejection [20][34]. However, the mechanism through which TIGIT blockade has an impact primarily on NK cells compared to T cells and the mechanism through which NK cells promote CD8+T cell responses need to be further elucidated, since these results are contradictory to those of previous studies, revealing the CD8+T or the Treg cell-mediated effect of TIGIT blockade using temporary depletion of these populations with specific antibodies [21][28][41][35,42,55]. A more recent study reported that anti-TIGIT mAbs enhanced IL-15-driven NK cell cytotoxicity in both B16F10 and LWT1 metastatic melanoma-bearing mice [55][69].
The potency of human anti-TIGIT blocking mAbs on CD8+T cells has been demonstrated in cancer patients. Cancer testis antigen NY-ESO-1-specific CD8+T cell responses are increased by the addition of blocking mAbs against TIGIT and/or PD-1 when peripheral blood mononuclear cells (PBMCs) from melanoma patients are stimulated with NY-ESO-1157–165 peptide. Furthermore, TIGIT blockade increases the capacity for proliferation and degranulation of CD8+TILs from advanced melanoma patients upon TCR stimulation using autologous non-CD3 cells and anti-CD3 mAbs [36][50]. Upon TCR stimulation with anti-CD2/anti-CD3/anti-CD28 microbeads, bone marrow (BM) CD8+T cells in MM patients show increased CD107a expression and cytokine production in response to TIGIT blockade [21][35]. When anti-TIGIT mAbs are added to ex vivo co-culture of CD3+TILs and Mel-624 cells expressing membrane-bound anti-CD3 scFv (Mel-624 OKT3), IFN-γ and IL-2 production by CD3+TILs from patients with endometrial, ovarian, kidney, head and neck, and lung cancers is promoted [18][14]. A recent study reported that antigen specific responses to CEF (CMV, EBV, flu) peptide are augmented by TIGIT blockade in peripheral blood CD8+T cells derived from pancreatic ductal adenocarcinoma (PDAC) patients after mFOLFIRINOX therapy [29][43].
3.3. Mode of Action of Anti-TIGIT Therapy
Competitive binding of TIGIT and CD226 to PVR has been known as a key mechanism of TIGIT-driven immune suppression, and anti-TIGIT blocking mAbs are presumed to reverse the suppression by inhibiting TIGIT binding to PVR. This may occur as a mode of action; however, several questions need to be addressed for its clinical success and further translation of other members of TIGIT family receptors into cancer immunotherapy.
-
Intracellular Regulation by Anti-TIGIT mAbs
Despite the importance of understanding the molecular interplay between TIGIT, CD226, and PVR, the mechanism through which extracellular signals from the receptor-ligand binding/receptor dynamics are integrated into the intracellular regulation, particularly in the context of anti-TIGIT therapy, remains unclear.
A recent study by Jin et al. reported that the effect of TIGIT blockade depends on tyrosine phosphorylation at Y322 of CD226, which was the first study to define the molecular requirements for anti-TIGIT blocking mAbs [29][43]. They showed that TIGIT-mediated intracellular inhibition of CD226 phosphorylation at Y322 was restored by TIGIT blockade. Moreover, CD226 mutant at Y322 (CD226Y322A) expressing CD8+T cells did not respond to TIGIT blockade, whereas CD226WT or CD226Y329A expressing CD8+T cells produced increased IFN-γ upon treatment with anti-TIGIT mAbs, suggesting that TIGIT blockade promotes T cell activation via CD226 phosphorylation at Y322 (Figure 2). CD226 dependent effect of anti-TIGIT mAbs was further shown in effector memory CD8+T cells expressing a low level of CD226 (CD226loCD8+Tem) not responsive to both antigen stimulation and anti-TIGIT mAb treatment. CD226 activation using anti-CD226 agonist mAbs renders CD226loCD8+Tem responsive to TIGIT blockade.
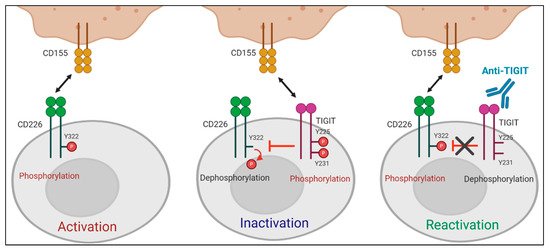
Figure 2. Role of CD226 in anti-TIGIT immunotherapy. TIGIT has a direct effect on intracellular regulation of CD226 activation in response to PVR binding. (Left) When TIGIT expression is absent or low, engagement of CD226 with PVR induces the phosphorylation of tyrosine 322 (Y322) on CD226, which leads to the activation of intracellular signaling cascades. (Middle) PVR preferentially binds to upregulated TIGIT over CD226. Upon interaction with PVR, the cytoplasmic tail of TIGIT is phosphorylated. This PVR-induced TIGIT phosphorylation inhibits T cell responses by promoting CD226 dephosphorylation. (Right) TIGIT blockade suppresses PVR-induced TIGIT phosphorylation and restores the impaired Y322 phosphorylation of CD226, thereby leading to T cell activation.
-
Isotype Selection of Anti-TIGIT mAbs
Recently, several studies have highlighted the importance of selecting appropriate fragment crystallizable (Fc) region for therapeutic antibodies. To date, the approved human therapeutic IgG antibodies belong to IgG1, IgG2, or IgG4 subclasses [56][70]. It is increasingly clear that binding of the Fc region of antibody to Fc gamma receptors (FcγRs) can elicit various immunomodulatory functions, including antibody dependent-cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-dependent cellular phagocytosis (ADCP) [57][71]. In addition, FcγR binding was reported to enhance agonistic activity of mAbs targeting tumor necrosis factor receptor superfamily members, such as CD28, CD137, CD40, and OX40 (CD134) [58][72].
The importance of the Fc domain of anti-TIGIT mAb is emphasized by the findings that anti-TIGIT mAb with Fc devoid of effector functions, which was intended to solely block TIGIT binding to its ligands, fails to exert any of anti-tumor efficacies in preclinical models [22][59][60][36,73,74]. It may be due to the loss of its depleting activity against TIGIT-expressing intratumoral Treg cells, which has been considered as a potential mechanism of anti-TIGIT mAb-mediated anti-tumor effect [60][74]; however, it is still not clear whether the anti-tumor efficacy of anti-TIGIT mAbs depends on Treg depletion, since there are recent reports that anti-TIGIT mAbs on mIgG2a isotype induce anti-tumor responses without evidence of Treg depletion in mouse tumor models [22][59][36,73]. It may possible that FcγR on APC could act as a scaffold to crosslink anti-TIGIT mAb bound to TIGIT on immune cells, which may enhance the effect of TIGIT antagonism independent of Treg cells. In addition, Han et al. recently have shown that the antibody-FcγR engagement induced activation of myeloid cells, leading to pro-inflammatory chemokine and cytokine secretion (Figure 3) [22][36]. Comparison of clinical activities of anti-human TIGIT mAbs with different Fc scaffolds could provide insight into whether FcγR binding is required for optimal anti-tumor responses of TIGIT blockades.
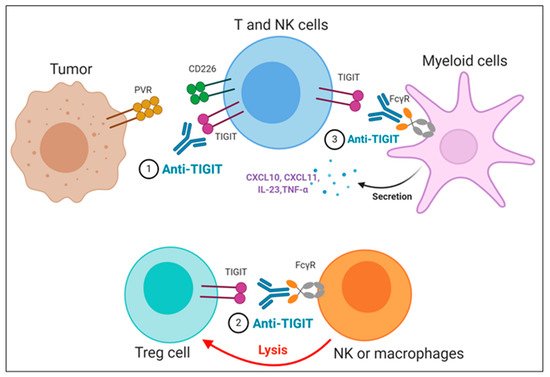
Figure 3. Proposed mechanisms of anti-TIGIT monoclonal antibodies (mAbs) in cancer immunotherapy. (1) Blockade of TIGIT could reverse the exhaustion of T and NK cell-mediated anti-tumor immunity. (2) Intratumoral regulatory T cells (Treg_) cells expressing high levels of TIGIT could be preferentially depleted by anti-TIGIT mAbs, presumably through antibody-dependent cellular phagocytosis (ADCP) by macrophages and/or antibody-dependent cellular cytotoxicity (ADCC) by NK cells. (3) The TIGIT mAb- fragment crystallizable (Fc) gamma receptors (FcγR) engagement could activate myeloid cells, leading to enhanced antigen presentation function and proinflammatory chemokine and cytokine secretion.
4. Anti-TIGIT Antibodies in Clinical Trials
Approximately 10 human anti-TIGIT mAbs, which have different IgG isotypes or mutant forms, have entered clinical trials. Table 1 summarizes publicly available data regarding antibody isotype, combination with different drugs, current development phase, and cancer types. Numerous clinical trials are evaluating the safety and the efficacy of anti-TIGIT mAb either as a monotherapy or in combination with PD-1/PD-L1 blockade or chemotherapies for the treatment of various cancers. Recently, the phase II CITYSCAPE trial presented significant response rates of tiragolumab plus atezolizumab in PD-L1-positive non-small cell lung cancer (NSCLC). The study revealed a significant objective response rate (ORR) improvement for the combination group (37% vs. 21%) as well as progression-free survival (PFS) improvement (5.6 vs. 3.9 months; hazard ratio [HR] 0.58). Importantly, patients in the combination group with high PD-L1 expression had an ORR of 66% compared with 24% in the atezolizumab group [61][75].
Table 1.
Clinical trials of TIGIT inhibitors.
| TIGIT Inhibitor | Sponsor | Isotype | Identifiers | Cancer Type | Combination | Phase | Recruitment Status | Start Date |
|---|---|---|---|---|---|---|---|---|
| ASP-8374 | Astellas Pharma Inc. | IgG4 | NCT03260322 | Advanced solid tumor | ASP-8374 alone; Pembrolizumab (anti-PD-1) |
Phase 1b | No longer recruiting | 8 September 2017 |
| NCT03945253 | Advanced solid tumor | ASP-8374 alone | Phase 1 | No longer recruiting | 5 August 2019 | |||
| BGB-A1217 | BeiGene Co Ltd. | IgG1 | NCT04047862 | Advanced solid tumor | Tislelizumab (anti-PD-1) | Phase 1 | Recruiting | 26 August 2019 |
| BMS-986207 | Bristol-Myers Squibb Co. | IgG1 (Fc receptor disabled) |
NCT02913313 | Advanced solid tumor | BMS-986207 alone; Nivolumab (anti-PD-1) |
Phase 1/2 | No longer recruiting | 29 November 2016 |
| NCT04150965 | Multiple myeloma | BMS-986207 alone; Dexamethasone+Pomalidomide |
Phase 1/2 | Recruiting | 16 April 2018 | |||
| NCT04570839 | Advanced solid tumor | COM-701 (PVRIG inhibitor) + Nivolumab (anti-PD-1) | Phase 1/2 | Recruiting | 31 August 2020 | |||
| NCT04065425 | Multiple myeloma | Dexamethasone + Pomalidomide | Phase 1/2 | Not yet recruiting | 1 October 2019 | |||
| COM-902 | Compugen Ltd. | IgG4 | NCT04354246 | Advanced solid tumor | COM-902 alone | Phase 1 | Recruiting | 31 March 2020 |
| AB154 (Domvanalimab) |
Arcus Biosciences Inc. | IgG1 (Fc receptor disabled) |
NCT03628677 | Advanced malignancy | AB154 alone; Zimberelimab (anti-PD-1) |
Phase 1 | Recruiting | 12 September 2018 |
| NCT04656535 | Recurrent Glioblastoma | Zimberelimab (anti-PD-1) | Phase 1 | Not yet recruiting | 31 January 2021 | |||
| NCT04262856 | PD-L1 positive lung cancer | Zimberelimab (anti-PD-1); Zimberelimab + etrumadenant (A2aR and A2bR antagonist) |
Phase 2 | Recruiting | 28 May 2020 | |||
| EOS-884448 | iTeos Therapeutics | IgG1 | NCT04335253 | Advanced tumor | EOS-884448 alone | Phase 1/2 | Recruiting | 18 February 2020 |
| Etigilimab (OMP-313M32) |
OncoMed | IgG1 | NCT03119428 | Advanced solid tumor | Etigilimab alone; Nivolumab (anti-PD-1) |
Phase 1 | Terminated | 2 May 2017 |
| IBI-939 | Innovent Biologics Inc. | Not disclosed | NCT04353830 | Advanced tumor | IBI-939 alone; Sintilimab (anti-PD-1) |
Phase 1a | Recruiting | 22 May 2020 |
| NCT04672356 | Advanced lung cancer | Sintilimab (anti-PD-1) | Phase 1a | Not yet recruiting | 28 January 2021 | |||
| NCT04672369 | Advanced NSCLC | Sintilimab (anti-PD-1) | Phase 1b | Not yet recruiting | 6 June 2021 | |||
| M-6223 | Serono Research Institute Inc, Merck KGaA | Not disclosed | NCT04457778 | Advanced solid tumor | M-6223 alone; Bintrafusp alfa (TGF beta ligand inhibitor) |
Phase 1 | Recruiting | 10 July 2020 |
| Vibostolimab (MK-7684) | Merck Sharp & Dohme Corp. | IgG1 | NCT02964013 | Advanced solid tumor | Vibostolimab alone; Pembrolizumab (anti-PD-1); Pembrolizumab + Pemetrexed + Carboplatin; Pembrolizumab + Carboplatin or Cisplatin + Etoposide |
Phase 1 | Recruiting | 13 December 2016 |
| NCT04305054 | Advanced melanoma | Pembrolizumab (anti-PD-1); | Phase 1/2 | Recruiting | 1 July 2020 | |||
| NCT04303169 | Melanoma | Pembrolizumab (anti-PD-1) | Phase 1/2 | Recruiting | 26 June 2020 | |||
| NCT04305041 | Refractory melanoma | Pembrolizumab + Quavonlimab (anti-CTLA4) | Phase 1/2 | Recruiting | 26 June 2020 | |||
| NCT04165070 | Advanced NSCLC | Pembrolizumab + Carboplatin + Paclitaxel; Pembrolizumab + Pemetrexed |
Phase 2 | Recruiting | 19 December 2019 | |||
| NCT02861573 | Prostate cancer | Pembrolizumab (anti-PD-1) | Phase 1/2 | Recruiting | 17 November 2016 | |||
| Tiragolumab (MTIG7192A) | Genentech Inc., Chugai Pharmaceutical Co. Ltd., Roche Holding AG | IgG1 | NCT04045028 | Relapse/Refractory Multiple myeloma and B-cell Non-Hodgkin lymphoma | Tiragolumab alone; Daratumumab (anti-CD38); Rituximab (anti-CD20) |
Phase 1 | Recruiting | 22 July 2019 |
| NCT02794571 | Metastatic solid tumor | Tiragolumab alone; Atezolizumab (anti-PD-L1); Chemotherapy (Carboplatin, Cisplatin, Etoposide, Paclitaxel, Pemetrexed) |
Phase 1 | Recruiting | 23 May 2016 | |||
| NCT03281369 | Metastatic esophageal cancer | Atezolizumab (anti-PD-L1); Atezolizumab + Cisplatin+5FU |
Phase 1/2 | Recruiting | 13 October 2017 | |||
| NCT04513925 | NSCLC | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 24 August 2020 | |||
| NCT04294810 | Metastatic NSCLC, PD-L1 selected | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 04 March 2020 | |||
| NCT04665843 | Metastatic head and neck cancer, PD-L1 positive | Atezolizumab (anti-PD-L1) | Phase 2 | Not yet recruiting | 21 January 2021 | |||
| NCT04543617 | Esophagus squamous cell carcinoma | Atezolizumab (anti-PD-L1) | Phase 3 | Recruiting | 28 September 2020 | |||
| NCT04300647 | Metastasis/Recurrent uterine cervix tumor, PD-L1 positive | Atezolizumab (anti-PD-L1) | Phase 2 | Recruiting | 30 June2020 | |||
| NCT03563716 | NSCLC, chemotherapy-naïve | Atezolizumab (anti-PD-L1) | Phase 2 | No longer recruiting | 10 August 2018 | |||
| NCT04665856 | Small-cell lung cancer | Atezolizumab + Carboplatin + Etoposide | Phase 3 | Recruiting | 4 January 2021 | |||
| NCT04619797 | Metastatic NSCLC | Atezolizumab + Pemetrexed + Carboplatin or Cisplatin | Phase 2 | Recruiting | 11 December 2020 | |||
| NCT04584112 | Triple-negative breast cancer | Atezolizumab + Nab-paclitaxel; Atezolizumab + Nab-pac-carbo-AC; Atezolizumab+Nab-pac-AC; |
Phase 1b | Recruiting | 28 September 2020 | |||
| NCT04256421 | Metastatic small-cell lung cancer | Atezolizumab + Carboplatin + Etoposide | Phase 3 | Recruiting | 4 February 2020 | |||
| NCT04540211 | Metastatic esophageal cancer | Atezolizumab + Paclitaxel + Cisplatin | Phase 3 | Recruiting | 4 November 2020 | |||
| NCT04524871 | Metastatic hepatocellular carcinoma | Atezolizumab + Bevacizumab (anti-VEGF) | Phase 1/2 | Recruiting | 2 November 2020 | |||
| NCT03869190 | Advanced urothelial carcinoma | Atezolizumab (anti-PD-L1) | Phase 1/2 | Recruiting | 1 June 2019 | |||
| NCT03193190 | Metastatic pancreatic ductal adenocarcinoma | Atezolizumab + Nab-Paclitaxe l+ Gemcitabine | Phase 1/2 | Recruiting | 5 July 2017 |
PD-L1: programmed death-ligand 1; NSCLC: non-small cell lung cancer; TGF: transforming growth factor; VEGF: vascular endothelial growth factor.