The development of molecular studies to define the somatic genetic alterations has revolutionized the diagnostic and therapeutic management of acute myeloid leukemia (AML). AML is a highly heterogenous disease that includes many molecular subtypes; each subtype is heterogeneous both for the presence of variable co-mutations and complex combinations of clones and subclones, changing during disease evolution and in response to treatment. The treatment of AML is changing from standardized schemes of induction and consolidation chemotherapy to tailored approaches according to molecular and genetic profiles and to targeted therapy. Several molecularly targeted therapies have been approved for the treatment of some AML patients, including mutation-specific targeted drugs such as FLT3, IDH1 and IDH2 inhibitors, mutation-independent targeted drugs such as the Bcl2 inhibitor venetoclax, the hedgehog inhibitor glasdegib and the CD33-targeted drug gemtuzumab ozogamicin. Furthermore, recent studies have shown the feasibility of a personalized medicine approach for the treatment of AML patients, where the therapy decisions are guided by the results of genomic studies.
- acute myeloid leukemia molecular classification prognosis diagnosis
1. Introduction
The development of massive parallel sequencing techniques has revolutionized the study of human cancers, allowing to sequence the entire genome and to provide detailed information on the genetic alterations present in tumor cells. The techniques of next generation sequencing (NGS) allowed to define the most recurrent genetic alterations observed in cancer cells, including gene mutations, small insertions/deletions (indels), gene fusions, alternative splicing and copy number alterations. NGS can provide, in a few days, the profile of genetic alterations in the blood or bone marrow samples from a patient with leukemia.
These dramatic progresses in the study of genomic alterations have considerably contributed to improve the understanding of the genetic alterations occurring in a heterogeneous disease, such as acute myeloid leukemia (AML). The current diagnostics of AMLs implies cytomorphology analysis, multiparameter flow cytometry, cytogenetics and molecular genetics. NGS studies have allowed to define the genomic landscape of AMLs, in its complexity and heterogeneity; ≥90% of AMLs display at least one gene mutation [1][2][3][4]. Different patterns of genetic instability are observed in AML cells; in fact, about 20% of AML patients can be defined according to fusion genes, 31% by chromosomal aneuploidies and 46% by gene mutations only [5]. Frequently, AML patients share mutations observed in normal subjects with clonal hematopoiesis; however, the majority of these patients acquired ≥two mutations, with clonal distribution [5]. The molecular classification of AMLs identified some major molecular subtypes: (i) AMLs characterized by peculiar translocation events (balanced rearrangements) leading to the formation of fusion genes and correspondent fusion proteins, including inv(16) with CBFB-MYH11, t(15;17) with PML-RARA, t(8;21) with RUN1-RUNXT1, inv(3) with GATA2-MECOM, MLL fusions and t(6;9) with DEK-NUP214; (ii) AMLs exhibiting chromatin-spliceosome gene abnormalities, including mutations of genes involved in RNA splicing (SRSF2, SF3B1, U2AF1, ZRSR2), chromatin (ASXL1) and transcription RUNX1); (iii) AMLs characterized by TP53 mutations, complex karyotype alterations and copy-number chromosome alterations; (iv) AMLs displaying mutations of the nucleophosmin 1 (NPM1) gene, mutually exclusive to other genomic rearrangements and with frequent co-mutations in hydroxymethylation genes (DNMT3A, TET2, IDH1, IDH2); (v) AMLs with chromosomal aneuploidies, characterized by double CEBPA mutation, with GATA2 and NRAS co-mutations found in about 30% of cases; (vi) AMLs with IDH2R172 mutation, defined as a distinct subgroup for the mutual exclusivity with NPM1 mutation and other class-defining lesions [3][4].
The application of NGS techniques has led to the identification of 40–50 genes recurrently mutated in AMLs [6][7][8]. Driver gene mutations play a key role in AML development with a clear pathogenetic and prognostic relevance [6][7][8]. The identification of these mutations has led to the individuation of molecular targets for mutation-based targeted therapy [9][10][11][12][13]. This progress has involved a careful definition of AML subtypes at the level of their main mutational events, of their genotypic and phenotypic heterogeneity, thus defining sensitive tools for risk stratification of these patients and for a sensitive and accurate evaluation of therapy response and for better planning the optimal treatment for each patient. Examples of treatment improvements are related to the development and to the clinical introduction of mutation-specific targeted small molecule inhibitors against mutant FLT3 or mutant IDH1/IDH2. In parallel, the introduction in the therapeutic armamentarium of venetoclax, a Bcl2 inhibitor, in association with hypomethylating agents is providing a consistent improvement in the survival of a part of older AML patients [9][10][11][12][13]. Thus, recently some new drugs were approved for the treatment of AML patients (Table I). Unfortunately, a significant proportion of patients develop resistance to these novel therapies whose molecular mechanism has been identified, in part bypassed by rationally designed combination therapies [9][10][11][12][13]. The final demonstration that these targeted treatments result in a clear benefit in terms of overall survival requires time and the careful definition of the most responsive AML patients.
2. The Fundamental Contribution of Precision Medicine to a More Rational and Predictive Risk Stratification of AML Patients
A correct risk stratification of AML patients is of fundamental importance for the adoption of the potentially optimal treatment strategy for each patient. Some clinical parameters and the integration of immunophenotypic characteristics, cytogenetic abnormalities and molecular mutations, co-occurring or in isolation, contributed to a more refined prognostic assessment.
The development of genomics has improved our understanding of AML development and resulted in novel modes of AML risk stratification that have been in part adopted in recently proposed classifications of AMLs. Prognostic risk of AMLs is defined at diagnosis according to the presence of specific cytogenetic and molecular aberrations [14][15][16]. Criteria for AML classification and risk stratification have been proposed by several organizations, including the European Leukemia NET (ELN) [14], National Comprehensive Cancer Network (NCCN) [15] and World Health Organization (WHO) [16]. The NCCN and ELN guidelines are the most adopted and stratify AML patients into three different risk groups: Favorable, intermediate and poor/adverse [14][15]. The most adopted risk classification is the 2017 ELN risk stratification; according to this classification, patients are classified into one of the three risk groups, including favorable, intermediate and adverse. Favorable prognosis group includes AMLs with acute promyelocytic leukemia (APL) t(15;17)(q22;q12), balanced translocations t(8;21)(q22;q22), biallelic mutated CEBPA and inv(16)(p13.1q22), mutated NPM1 without FLT3-ITD or with FLT3-ITDlow. The intermediate group comprises mutated NPM1 with FLT3-ITDhigh, WT-NPM1 without or with FLT3-ITDlow, t(9;11), MLLT3-MLL and cytogenetic abnormalities neither favorable or adverse. The adverse AML group comprises AMLs with complex karyotype, inv(3)(q21q26)/t(3;3)(q21;q26), DEK-NUP214 t(6;9)(p23;q34), RPN1-EVI1, t(6;11), −5 or del(5q), −7 or abnormal (17p) or monosomal karyotype, TP53 mutations, RUNX1 mutations, ASXL1 mutations, FLT3-ITDhigh isolated without NPM1 mutations and with normal karyotype. The NCCN and ENL adopt a similar classification scheme for favorable-risk AMLs, although the criteria for favorable risk differ in some respects in these two evaluation systems [14][15]. It is important to point out that each of these risk groups is consistently heterogeneous, even considering the favorable-risk AML group. Thus, a consistent genotypic and clinical heterogeneity exists within the favorable risk AML, a variability observed also in single molecularly-defined AML subtypes.
A recent study by Herold and coworkers, on 1116 adult AML patients not selected by genetics, validated the ELN-2017 classification and showed that: (i) In 599 patients < 60 years, the overall survival (OS) was 64% for ELN-2017 favorable, 42% for intermediate-risk and 20% for adverse-risk AMLs; (ii) In 517 patients > 60 years, corresponding five-year overall survival (OS) was 37%, 16% and 6% [17]. The analysis of the mutational profile showed that the large majority of RUNX1, ASXL1 and TP53 mutations were observed in the adverse risk group; SRSF2 and BCOR mutations were more frequent in the adverse group than in the two other groups; MLL-PTD mutations were more frequent in the adverse and intermediate groups, compared to the favorable group; NRAS and DNMT3A mutations were more frequent in the favorable-intermediate groups compared to the adverse group [17]. These authors proposed to refine the 2017 ELN classification by separating a very favorable subgroup (patients with inv(16)/t(16;16) or biallelic CEBPA mutations) from the favorable group, and a very adverse subgroup (patients with TP53 mutations and a complex karyotype) from the adverse group [17].
The 2017 ELN stratification system has provided and continues to provide an essential support to the risk evaluation of AML patients both in clinical current practice and in clinical trials. However, it is evident that 2017 ELN cannot predict the real risk of a part of AML patients, resulting in either an underestimation or in an overestimation of the individual patient’s risk. Furthermore, the 2017 ELN is based on scoring systems that are intrinsically limited by significant heterogeneity existing in AML subtypes. Several recent studies have provided evidence that the 2017 ELN classification can be implemented through a better evaluation of the impact of the individual AML mutational profile; only a technology like NGS makes it feasible to capture the genetic heterogeneity underlying AML heterogeneity, at individual level.
Risk stratification systems integrating mutational or gene expression data were found to add prognostic value to the current ELN risk classification [18][19]. Risk classification of AML based on a combination of molecular and clinical data may contribute to improve AML patient stratification. An example of this approach is given by the prognostic model for AML patients recently proposed by Ma and coworkers; in this model, several parameters including age, hematopoietic cell transplantation-comorbidity index, white blood cell count, hemoglobin, biallelic CEBPA, DNMT3A mutations, FLT3-ITD/NPM1 status and ELN cytogenetic risk status were identified as independent prognostic factors for overall survival in multivariate analysis [20]. This model showed a good performance with a C-index of 0.74 and can be applied to both young and older AML patients, and allows also the distinguishing of eligible candidates for hematopoietic stem cell transplantation [20].
3. The Contribution of the Machine Learning Approach to Improve the Assessment of AML Diagnosis and Prognosis
Machine learning is a branch of computer science and statistics that represents a form of artificial intelligence, based on the development of predictive and descriptive models by learning from training data rather than being pre-programmed according to rigid schemes; the learning approach implies both supervised learning and an unsupervised learning [21][22]. Therefore, machine learning can be considered as a form of interpretation and analysis of a specific reality based on the accumulation and elaboration of thousands of data, allowing the development of algorithms suitable to analyze the individual complexity and heterogeneity. Thus, it is not surprising that machine learning has rapidly found many applications in medicine from diagnosis, to prognosis and treatment [23]. The applications include also the management of hematological malignancies and particularly of AMLs, at the level of the analysis of genomic and gene expression data for diagnostic, prognostic and therapeutic purposes [21][22][24].
Various recent studies have shown the support of a machine learning approach to the analysis of genomic and transcriptomic data on AML samples.
Some studies were focused to explore a machine learning approach based on large dataset of mutational profiles and clinical data to perform diagnosis of several bone marrow myeloid neoplasms [25] and to predict the outcomes of myelodysplasias, myeloproliferative disorders and chronic myelomonocytic leukemia, particularly for that concerns the risk of AML transformation [26]. Radakovich et al. have used a machine learning approach to explore the genotype-phenotype correlations in patients with MDS and related myeloid malignancies using a large genomic database based on 2697 patients [27]. This analysis showed some associations between genotype and clinical phenotype: SF3B1 mutations were associated with normal karyotype and some clinical features and TP53 mutations were associated with complex karyotype; clinical characteristics were also associated with specific genomic alterations: Normal karyotype correlated with the presence of SF3B1, ZRSR2 and DNMT3A mutations and absence of TP53, ASXL1 and KRAS mutations, while age <65 years was associated with the presence of NRAS and JAK2 mutations and the absence of TET2, SF3B1 and SRSF2 mutations [27]. These observations support the existence of a link between mutational data and clinical characteristics [27].
Many studies showed the consistent impact of machine learning approach in the discovery of algorithms to improve the AML classification, prognosis prediction and outcomes and screening of drug sensitivity.
Gerstung et al., through the analysis of 1540 AML patients with available matched genomic-clinical data (knowledge bank), developed multistage statistical models more accurately predicting likelihoods of remission, relapse and mortality [24]. This study was based on a modified regression-based method for estimating the likelihood of survival whether a patient received hematopoietic stem cell transplantation in first remission or after relapse [28]. Particularly, this study showed that: (i) Clinical and demographic factors, such as patient age, performance status and blood counts, exerted the most influence on early death rates, including death in remission (due to treatment-related mortality); (ii) genomic features, mostly influencing the dynamics of disease remission and relapse [24]. Using a knowledge bank to model patient outcomes, a substantial fraction (about 1/3) was reclassified and would have their treatment altered compared to current recommendations [28]. Furthermore, personal tailored management decisions could reduce the number of hematopoietic stem cell transplants by 20–25%, while maintaining overall survival rates [28]. Furthermore, about 15% of ELN favorable risk patients are predicted to potentially benefit from stem cell transplantation in first complete remission [28]. It is important to point out that the accuracy of the predictive potential of knowledge bank-based systems largely depend on continuously updated databases based on thousands of patients [28].
Recently, Fleming and coworkers proposed a machine learning (ML) approach to develop a hierarchical prognostic risk model that hierarchically categorizes cytogenetic and molecular factors into groupings that accurately predict survival [25]. This approach was used to explore two large cohorts of AML patients that were classified into four prognostic groups: good (30%), intermediate (26%), poor (26%) and very poor (18%); the ELN2017 classification evaluated these AMLs as: good (39%), intermediate (31%) and poor (30%) [29]. It is important to note that, in this system of AML prognostication, a large number of molecular parameters was taken in account: Complex karyotype, inv(16), CEBPAdmut, inv(3)/t(3;3), FLT3-ITD, spliceosome mutations (U2AF1, SRSF2 or SF3B1), NPM1mut (in the absence of FLT3-ITD), t(8;21), MLL translocations, NRASmut, TP53mut, ASXL1mut [29]. This evaluation system allowed the prognostication of many AML subgroups: (i) In the group characterized by complex karyotype, the presence of high-risk monosomies or chromosomal abnormalities or TP53 mutations have a very poor prognosis, whereas complex karyotypes without these alterations have a better prognosis; (ii) CEBPAdmut AMLs have a good prognosis, particularly when associated with NRAS mutations; (iii) co-occurrence of FLT3-ITD and spliceosome mutations was associated with very negative outcome; (iv) FLT3-ITD high allelic ratio (>0.5) has a very poor prognosis when present in the absence of concomitant NPM1 mutations; (v) triple mutant NPM1/DNMT3A/FLT3-ITD AMLs display a poor prognosis; (vi) AMLs with spliceosome mutations display a poor prognosis when associated with ASXL1 mutations or ASXL1 heterozygous deletion; (vii) among NPM1-mutant AMLs, NRAS co-mutations identified a subgroup associated with good prognosis, whereas those associated with IDH1 mutations display an intermediate prognosis; (vii) the presence of KIT mutations in t(8;21) AMLs was associated with an intermediate prognosis [29].
Using a machine learning approach, Shreve et al. have developed a novel prognostic model of AMLs that incorporates clinical, cytogenetic and mutational data to predict personalized outcomes specific of individual patients [26]. This study was based on genomic data from 3421 patients; a machine learning algorithm capable of accounting for survival was used to generate the new model: In this model, clinical and molecular variables were randomly selected for inclusion in determining overall survival [30]. The analysis of the mutational impact in the various cytogenetic risk groups showed that the most frequently mutated genes in low risk were NRAS, KIT, FLT3 and KRAS, in intermediate risk were NPM1, FLT3, DNMT3A, IDH2, TET2 and in high risk were TP53, DNMT3A, NRAS, RUNX1, PTPN11 [30] (Figure 1). Importantly, when assayed on individual data from four cohorts of patients, this evaluation system showed a C-index for overall survival ranging from 0.80 to 0.85 compared to 0.59 when using the 2017 ELN criteria [30]. This study reached also another important conclusion concerning the differential impact of genomic alterations on overall survival in each cytogenetic risk group, thus indicating all the complexities relative to the incorporation of mutational data into risk stratification [30].
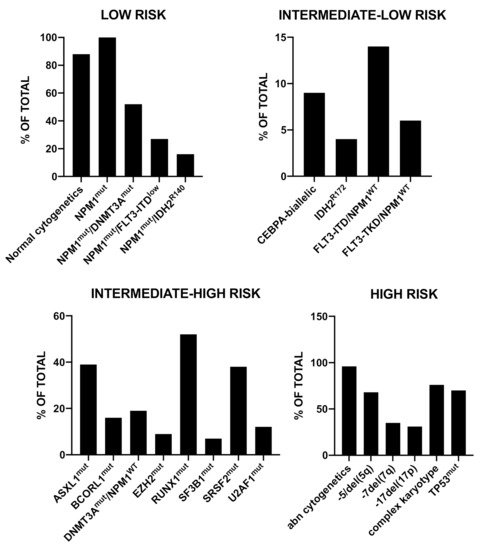
Figure 1. Recurrent gene mutations observed in a group of 3421 AML patients subdivided into three prognostic risk groups (low risk, intermediate risk and high risk), according to a machine learning algorithm accounting for survival. The data are reported in Shreve et al., 2019 [30]. Abbreviations: mut: mutant; del: deletion; abn: abnormal.
Awada et al. have used the machine learning approach to improve the subclassification and prognostication of AML through the collection and analysis of genomic data from a multicenter cohort of 6788 AML patients [31]. Using a logistic regression model some mutations resulted enriched in pAML (CEBPAmono, CEBPAbiallelic, DNMT3A, FLT3-ITD, FLT3-TKD, GATA2, IDH1, IDH2R140, NRAS, NPM1, WT1) and other mutations in sAML (ASXL1, RUNX1, SF3B1, SRSF2, U2AF1, −5/del(5q), −7/del(7q), −17/del(17P), del(20q), +8 and complex karyotype) [31]. Using the Bayes latent class analysis, four unique genomic clusters of distinct prognoses were identified: low risk, intermediate-low risk, intermediate-high risk and high risk [27]. The generation of a random forest model allowed the extraction of invariant genomic features driving each group; the main genomic alterations observed in these four groups are reported in Figure 2 [31].
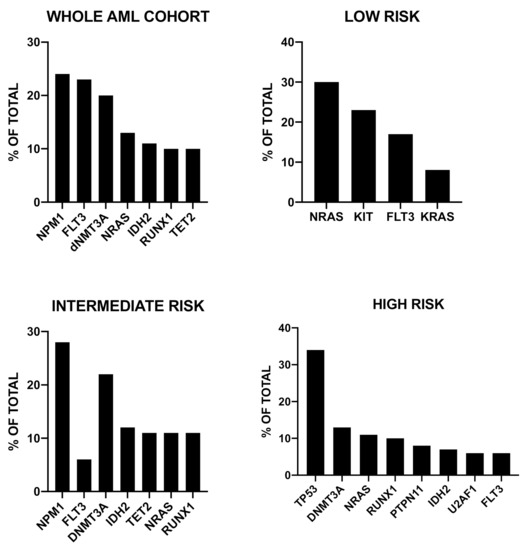
Figure 2. Recurrent genetic abnormalities observed in 6788 AML patients stratified into four prognostic risk groups (low risk, intermediate-low risk, intermediate-high risk and high risk), according to a machine learning algorithm. The data are reported in Awada et al., 2020 [31].
Siddiqui et al. have shown the potential of machine learning algorithms, trained using factors available at the time of admission for AML treatment, to predict death during the patient’s hospitalization; this study was based on the acquisition of data relative to a total of 29,613 patients with AML [32].
The machine learning approach was shown to be very useful for a prognostic identification of AMLs with specific driver mutations. Supervised machine learning analysis of the profile of genetic alterations observed patients with RUNX1-RUNXT1 AMLs identified the presence of concurrent NRAS mutations and the absence of mutations in ASXL2, RAD21, KIT and FLT3 genes and low mutational burden as conditions of favorable genetic risk [33]. According to these data, the patients were stratified into a poor genetic risk group associated with lower overall survival and relapse-free survival, compared to the group of patients classified as good genetic risk [33]. In another study, Patkar and coworkers, using the machine learning approach, have developed a scoring model for the risk stratification of AML patients with NPM1 mutations; in this model, the five top variables to predict the outcomes of these leukemias were NPM1 VAF (variant allelic frequency), FLT3-ITD VAF, presence of IDH2 mutations, DNMT3A R882 mutation, and type A NPM1 mutation: The presence of type A NPM1 and IDH2 mutations and low levels of FLT3-ITD VAF and NPM1 VAF and absence of DNMT3A mutations were favorable prognostic factors [34]. This scoring system allowed to stratify NPM1-mutant AMLs into three groups with favorable, intermediate and poor genetic risk, exhibiting a remarkably different outcome in terms of overall survival and relapse-free survival. Furthermore, a strong statistical correlation was observed between ML-derived genetic risk and post-induction flow cytometry minimal residual disease [34].
The analysis of gene expression profiling was essential to improve the molecular classification of AMLs and to identify new biomarkers suitable for clinical studies. Initial studies have led to the identification of gene expression signatures with a prognostic predictive value. Thus, Bullinger et al. reported the identification of a 133-gene clinical-outcome predictor, which accurately predicted overall survival, including also patients with AML with a normal karyotype [35]. Li et al., through the analysis of a very large set of data derived from different cohorts of AML patients, identified a robust prognostic signature composed of 24 genes, capable of predicting overall survival and event-free survival. Furthermore, this signature provides a significant improvement of the ELN risk classification of AML [36]. Marcucci et al., through an epigenetic analysis involving the analysis of genes with differently methylated regions in older AML patients, reported the identification of seven genes whose expression was associated with outcome: A low score was associated with a better complete remission rate and longer disease-free survival and overall survival [37]. Ng et al. used an approach aiming to develop predictive and/or prognostic biomarkers related to stemness, based on the identification of genes that are differentially expressed between leukemic stem cell-positive and leukemic stem cell-negative cell fractions derived from 78 AMLs. Using this approach, they identified 17 genes whose expression score was highly prognostic in five different cohorts of AML patients, comprising different AML subtypes [38]. More recently, some machine learning studies were based on the analysis of transcriptomic AML profiles. Warnat-Herresthal reported the use of LASSO (Least Absolute Shrinkage and Selection Operator) regression analysis, a machine learning method allowing to automatically select the most predictive characteristics, for the automatic diagnosis and classification of AMLs based on transcriptomic data [35]. This analysis utilizes data from 105 studies, involving 12,029 AML, ALL (Acute Lymphoblastic Leukemia), MDS patients and normal subjects; this system identified AMLs based on transcriptomic data, with >99% accuracy [39]. Interestingly, Wagner et al. used an artificial neural network (ANN)-based machine learning approach to a dataset of 593 AML and identified a three-gene expression signature comprising CALCRL, CD109 and LSP1, predictive of both overall and relapse-free survival [40]. This three-gene prognostic index separated the adult AML patients in each 2017 ELN cytogenetic risk category into subgroups with different survival probabilities and allowed also the identification of patients with high-risk features [40]. The prognostic impact of this three-gene index was validated in different cohorts of AML patients, including childhood AML [40]. In another study, Rouphail and coworkers have used a machine learning approach, based on the analysis of transcription data relative to 242 AML patients, mainly NPM1-mutated, to define differences in transcription signatures of NPM1mut/FLT3-ITD compared to NPM1mut/FLT3-WT [41]. The algorithm that this study developed, identified 20 genes that are highly specific for NPM1mut/FLT3-ITD AMLs; these genes affect key biochemical pathways involved in the regulation of cell differentiation, proliferation, mitochondrial oxidative phosphorylation, histone modification and lipid metabolism [41].
Other few recent studies have started to explore the possible contribution of machine learning approach to develop models to predict the outcome of allogeneic stem cell transplantation. Thus, Gandelman and coworkers provided preliminary evidence that machine learning computational studies may better reveal biomarkers and stratify risk of patients with hematological malignancies undergoing allogeneic SCT, than the current approach based on cumulative severity [42]. Choi and coworkers have shown the feasibility of the machine learning-based approach, using random forest models, to predict survival after allogeneic stem cell transplantation in hematologic malignancies [43]. Finally, Nazha and coworkers have developed a personalized prediction model for outcomes after allogeneic SCT in patients with MDSs; the algorithm developed in this model identified the following variable prior to cell transplant impacting overall survival: Age, TP53, RAS, JAK2, ZRS2 and CUX1 mutations, cytogenetic profile, conditioning regimen, donor age, WBC count, hemoglobin and diagnosis of therapy-related MDS [44]. Importantly, this novel model is able to provide survival probability at different time points specifically for a given patient [44].
A machine learning-based approach was used also in drug discovery and development. Thus, Lee et al. have reported the development of a novel method for predicting AML drug sensitivities; this approach incorporates information learned from the Tumor Cancer Genome Atlas to help to support the link between genetic alterations and the pattern of drug sensitivity [45]. Using this approach, the authors identified SMARCA4 as a marker and driver of sensitivity to the topoisomerase II inhibitors mitoxantrone and etoposide, showing that the increased sensitivity predicted by the model was confirmed by in vitro assays [41]. Other studies reported the use of machine learning in drug screening: Chen et al. adopted machine learning to evaluate potential STAT3 inhibitors in AML [46]; Cutler and Fridman developed a machine learning-based model to predict high sensitivity to the compound FLX925, a FLT3 inhibitor, in AML [47].
Machine learning algorithms have been also applied to the detection and analysis of minimal residual disease. Minimal (or measurable) residual disease refers to the detection of residual leukemic cells below the threshold for morphological recognition and represents an important tool to evaluate the response after anti-leukemia treatment and is an important prognostic indicator for AML. MRD (Minimal Residua Disease) can be detected either using molecular assays or multiparameter flow cytometry (MFC). MFC has been and is extensively used in the detection of MRD in various hematological malignancies, including AML and MDS. However, the current methodology of MFC is complex at the level of data interpretation for the problem of manual flow cytometer gating. To bypass these difficulties, Ko et al. have used two machine learning techniques to develop an MFC interpretation algorithm for MRD detection using two large cohorts of AML and MDS patients [48]. High clinical validity of the algorithm was demonstrated through appropriate outcome prediction in the post-induction chemotherapy setting [48].
4. An Integrated Approach Is Required for the Development of Personalized Medicine in AML
The diagnosis of AMLs requires a multidisciplinary, integrated diagnostic approach, based on cytomorphology, cytochemistry, immunophenotyping, cytometry and molecular genetics [49]. A multidisciplinary diagnostic approach is today fundamental for appropriate AML subtype identification, patient prognostication and to define optimal therapy and also for definition of markers to monitor response to therapy [49]. This approach is particularly important in view of the development of precision medicine.
Furthermore, functional approaches, such as ex vivo drug sensitivity and resistance profiling, may cooperate with genomic, epigenomic and transcriptomic data in the identification of new targeted therapies and thus increase the number of drugs that can be tailored to AML patients [50].
Tyner and coworkers performed an integrative analysis of clinical, cytogenetic, molecular genetic and transcriptional data, together with in vitro testing of primary samples, examining drug sensitivity against 122 different compounds [46]. This functional genomic analysis was performed on a large cohort of 562 AML patients based on whole exome sequencing, RNA-sequencing and ex vivo drug sensitivity analyses [51]. This approach showed several relevant findings: (i) Genetic subgroups, including TP53 or ASXL1 mutations, were associated with widespread drug sensitivity; (ii) a sensitivity of FLT3-ITD mutant AMLs to FLT3 inhibitors; (iii) NRAS-mutant AMLs resistant to most of the drugs, but sensitive to MAPK inhibitors; (iv) IDH2-mutant AMLs are sensitive to several drugs, whereas the contrary is true for IDH1-mutant AMLs; (v) RUNX1-mutant AMLs are sensitive to PIK3C/MTOR inhibitors; (vi) AMLs with mutations of spliceosome genes display a peculiar pattern of drug sensitivity; (vii) triple mutant NPM1/FLT3/DNMT3A AMLs are sensitive to ibrutinib [51]. Co-occurrence of some genetic mutations and some gene expression clusters were associated with and predicted response to specific drugs [51].
Recent advancements in understanding of the molecular alterations of AMLs have determined the generation of a growing number of molecularly targeted drugs, such as FLT3 and IDH inhibitors. However, several limiting factors hinder the development of effective single-agent targeted therapies, including the more or less pronounced heterogeneity of AML subtypes, the emergence or amplification of pre-existing subclones leading to relapse, and protective signals mediated by the tumor microenvironment. The combination of drugs that target different pathways may represent a valuable strategy to improve the response and to reduce the resistance mechanisms by tumor cells. Thus Kurtz et al. have evaluated, on fresh AML blast cells, the sensitivities to combinations of molecularly targeted drugs acting on different cell-signaling responses, and have correlated these responses with the diagnostic clinical/genetic/cytogenetic and cellular features of the various patient samples [52]. These studies showed that, for AML cells, several combinations of targeted agents that include venetoclax (a Bcl2 inhibitor) and a kinase inhibitor are affective [52].
Various authors have reported automated systems for ex-vivo drug testing capable of predicting chemosensitivity in AML patients [53][54][55].
Other studies have evaluated the chemogenomic landscape of molecularly-defined AML subsets. Thus, Simon et al. have evaluated, in parallel, the mutational spectrum and gene expression profile of RUNX1-mutated AMLs and have correlated these results to drug sensitivity assayed in vitro [56]. Chemical screening showed that most RUNX1-mutated AML specimens are sensitive to glucocorticoids, resulting in an inhibitory effect on cell proliferation [56]. Moison et al. have reported a comprehensive genomic and transcriptomic analysis of a cohort of AML patients with complex karyotype and identified in these AMLs the frequent (about 80%) aberrant expression of the HMGA2 oncogene, in a TP53-independent manner [46]. HMGA2 mediated sensitization of complex karyotype AMLs to G2/M checkpoint, thus offering a potential therapeutic opportunity using drugs such as CHK1 and PLK1 inhibitors [57].
As above reported, venetoclax-based therapy (venetoclax in association with hypomethylating agents such as azacytidine or decitabine) can induce responses in about 70% of older previously untreated AML patients. However, upfront resistance, as well as acquired resistance determining relapse limit the effectiveness of this treatment. Zhang and coworkers have used an integrated genomic and functional screen data analysis to identify biomarkers predicting venetoclax sensitivity and resistance in AML and to identify venetoclax combination strategies to bypass resistance mechanisms [54][55]. By integrating the clinical data, exome and RNA sequencing, and inhibitor data from samples derived from approximately 200 samples of treated patients, several conclusions were reached: A myelomonocytic phenotype of leukemic cells (as supported by high CD14 expression), upregulation of BCL2A1 and CLEC7A and mutations of PTPN11 and KRAS were associated with resistance to venetoclax and multiple venetoclax combinations; venetoclax in combination with an inhibitor of the antiapoptotic protein MCL1 (AZD5991) induced synthetic lethality and bypassed venetoclax resistance [58][59].
Another study confirmed a link between PTPN11 mutations and venetoclax resistance and showed also that mutant PTPN11 induces, in leukemic cells, an increase of oxidative phosphorylation and glycolysis: This metabolic modification determines resistance to venetoclax that can be bypassed by a MCL1 inhibitor [60]. Recent studies have characterized AMLs bearing PTPN11 mutations showing several peculiar findings: Frequent myelo-monocytic morphology; frequently co-mutated with NPM1 and FLT3-ITD and less frequently with IDH2 and complex karyotype; an adverse prognosis compared to PTPN11-WT AMLs (8.4 vs. 13.6 months of median overall survival) [61].
Another study confirmed the resistance to venetoclax of monocytic AMLs: These leukemic cells have a distinct transcriptomic profile, lose expression of BCL2 and rely on MCL1 to mediate oxidative phosphorylation and survival [62].
Spinner et al. used an ex-vivo drug screening to define novel drug sensitivity patterns for informing personalized therapy in a group of 21 MDS patients resistant to hypomethylating agents [63]. Ex-vivo drug screening was performed within a clinically actionable time frame (a median time of 15 days) and showed drug sensitivity patterns heterogeneous, defining distinct patient clusters with differential sensitivity to hypomethylating agents, anthracyclines, histone deacetylase inhibitors and kinase inhibitors. Furthermore, a synergy between hypomethylating agents and venetoclax was observed [63]. These results on drug sensitivity informed personalized therapy. In 21 patients with ex vivo and in vivo clinical response data, the ex-vivo drug sensitivity screening platform showed a positive predictive value of 0.92, negative predictive value of 0.82, and overall accuracy of 0.85 [63].
While the above reported studies supported a role of integrated chemogenomic approach to identify AML subsets associated with sensitivity/resistance to specific drugs or to identify new potential treatments in AML subtypes, other recent studies directly implied the chemogenomic approach into a clinical trial.
Snijder et al. evaluated, in a prospective study, the feasibility and efficacy of ex-vivo drug-response profiling to guide personalized treatment selection across large panels of possible treatments for patients affected by aggressive hematological malignancies, including AMLs [64]. This study was based on a new image-based, drug-response profiling technique called pharmacoscopy, which uses high-throughput, automated confocal microscopy, immunofluorescence and singe-cell image analysis [64]. Pharmacoscopy, retrospectively predicted the clinical response of 20 AML patients to induction therapy with 88% accuracy [64]. Seventeen patients received the pharmacoscopy-guided treatment, providing preliminary evidence that this treatment is feasible, safe and effective [64].
In another study, Collignon et al. have assessed the feasibility of a tailored treatment strategy guided by systematic ex vivo drug sensitivity/resistance profiling and targeted NGS for patients with refractory/relapsed AML [65]. A tailored treatment strategy could be achieved in 47/55 AML patients: Five based only on targeted NGS, six on drug sensitivity/resistance profiling and 36 on both techniques [65]. The tailored treatment strategy was available in <21 days for 28 patients participating to the study; three to four potentially active drugs were selected for each patient; five patients resulted resistant to the whole panel of drugs tested [65]. Seventeen patients received a tailored treatment strategy and resulted in 4 complete remissions, one partial remission and five decreased peripheral blast cell counts [65].
References
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mingall, A.J.; Robertson, A.G.; Hoadley, A.S.K.; Triche, T.J.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 20959–22074.
- Hou, H.A.; Lin, C.C.; Chou, W.C.; Liu, C.Y.; Chen, C.Y.; Tang, J.L.; Lai, Y.J.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia 2014, 28, 50–58.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, K.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Hauser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of acute myeloid leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934–946.
- Moarii, M.; Papaemmanuil, E. Classification and risk assessment in AML: Integrating cytogenetics and molecular profiling. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 8, 37–44.
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 9–57.
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; DiNardo, C.D.; Daver, N. Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 2020, 10, 506–525.
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New directions for emerging therapies in acute myeloid leukemia: The next chapter. Blood Cancer J. 2020, 10, 107.
- Yu, J.; Jiang, P.; Sun, H.; Zhang, X.; Jiang, Z.; Li, Y.; Song, Y. Advances in targeted therapy for acute myeloid leukemia. Biomarker Res. 2020, 8, 17.
- Samra, B.; Knopleva, M.; Isidori, A.; Daver, N.; DiNardo, C. Venetoclax-based combinations in acute myeloid leukemia: Current evidence and future directions. Front. Oncol. 2020, 10, 562558.
- Ganget, N.; Tefferi, A. Venetoclax-based chemotherapy in acute and chronic myeloid neoplasms: Literature survey and practice points. Blood Cancer J. 2020, in press.
- Marando, L.; Huntly, B.J.P. Molecular landscape of acute myeloid leukemia: Prognostic and therapeutic implications. Curr. Oncol. Rep. 2020, 22, 61.
- Hou, H.A.; Tien, H.F. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020, 27, 81.
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447.
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A. Acute myeloid leukemia, version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 926–957.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiede, J.; Borowitz, M.J.; Le Beau, M.M. The 2016 revision in the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 1391–2405.
- Herold, T.; Rothenberg-Thurley, M.; Grumwald, V.V.; Janke, H.; Goerlich, D.; Sauerland, M.S.; Kostandin, N.P.; Dufour, A.; Schneider, S.; Neusser, M.; et al. Validation and refinement of the revised 2017 European Leukemia Net genetic risk stratification of acute myeloid leukemia. Leukemia 2020, in press.
- Wang, M.; Lindberg, J.; Kelvebring, D.; Nillsson, C.; Mer, A.S.; Rantalaien, M.; Lehmann, S.; Gronberg, H. Validation of risk stratification models in acute myeloid leukemia using sequencing-based molecular profiling. Leukemia 2017, 31, 2029–2036.
- Wang, M.; Lindberg, J.; Klevebring, D.; Nillsson, C.; Lehmann, S.; Gronberg, H.; Rantalainen, M. Development and validation of a novel RNA sequencing-based prognostic score for acute myeloid leukemia. J. Natl. Cancer Inst. 2018, 110, 1094–1101.
- Ma, T.T.; Lin, X.J.; Cheng, W.Y.; Xue, Q.; Wang, S.Y.; Liu, F.J.; Yan, H.; Zhu, Y.M.; Shen, Y. Development and validation of a prognostic model for adult patients with acute myeloid leukemia. EBioMedicine 2020, 62, 103126.
- Radakovich, N.; Nagy, M.; Nazha, A. Machine learning in haematological malignancies. Lancet Haematol. 2020, 7, e541–e550.
- Radakovich, N.; Cortese, M.; Nazha, A. Acute myeloid leukemia and artificial intelligence, algorithms and new scores. Best Pract. Res. Clin. Hemat. 2020, 33, 101192.
- Goecks, J.; Jalili, V.; Heiser, L.M.; Gray, J.W. How machine learning will transform biomedicine. Cell 2020, 181, 92–101.
- Eckardt, J.N.; Bornhauser, M.; Wendt, K.; Middeke, J.M. Application of machine learning in the management of acute myeloid leukemic: Current practice and future prospects. Blood Adv. 2020, 4, 6077–6085.
- Beau Hilton, C.; Meggendorfer, M.; Sekeres, M.A.; Shreve, J.; Radakovich, N.; Rouphail, Y.; Walter, W.; Hutter, S.; Padron, E.; Savona, M.R.; et al. Geno-clinical model for the diagnosis of bone marrow myeloid neoplasms. Blood 2019, 134 (Suppl. S1), 4238.
- Morita, K.; Wang, F.; Makishima, H.; Yan, Y.; Yoshizato, T.; Yoshida, K.; Przychodzen, B.P.; Patel, K.; Bueso-Ramos, C.E.; Gumbs, C.; et al. Pan-myeloid leukemia analysis: Machine learning-based approach to predict phenotype and clinical outcomes using mutation data. Blood 2018, 132 (Suppl. 1), 1801.
- Radakovich, N.; Malcovati, L.; Meggendorfer, M.; Sekeres, M.A.; Shreve, J.; Beau Hilton, C.; Rouphail, Y.; Walter, W.; Hutter, S.; Gallì, A.; et al. Henotype-phenotype correlations in patients with myeloid malignancies using explainable artificial intelligence. Blood 2020, 138 (Suppl. S1), 31–32.
- Gerstung, M.; Papaemmanuil, E.; Martincorena, I.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Heuser, M.; Thol, F.; Bolli, N.; Ganly, P.; et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat. Genet. 2017, 49, 332–340.
- Fleming, S.; Tsai, C.H.; Dohner, H.; Dohner, K.; Papaemmanuil, E.; Tien, H.F.; Reynolds, J.; Wei, A.H.; Hou, H.A. Use of machine-learning in 2074 cases of acute myeloid leukemia for genetic risk profiling. Blood 2019, 134 (Suppl. S1), 1392.
- Shreve, J.; Meggendorfer, M.; Awada, H.; Mukherjee, S.; Walter, W.; Hutter, S.; Makhoul, A.; Beau Hilton, C.; Radakovich, N.; Nagata, Y.; et al. A personalized prediction model to risk stratify patients with acute myeloid leukemia (AML) using artificial intelligence. Blood 2019, 134 (Suppl. S1), 2091.
- Awada, H.; Durmaz, A.; Gurnari, C.; Kishtagari, A.; Meggendorfer, M.; Kerr, C.M.; Kuzmanoviuc, T.; Durrani, J.; Nagata, Y.; Rdivoyevitch, T.; et al. The application of machine learning to improve the subclassification and prognostication of acute myeloid leukemia. Blood 2020, 136 (Suppl. S1), 28.
- Siddiqui, N.S.; Klein, A.; Godara, A.; Varga, C.; Buchsbaum, R.J.; Hughes, M.C. Supervised machine learning algorithms using patient related factors to predict in-hospital mortality following acute myeloid leukemia therapy. Blood 2019, 134 (Suppl. S1), 3435.
- Shaikh, A.F.; Kakirde, C.; Dhamne, C.; Bhanshe, P.; Joshi, S.; Chaudhary, S.; Chatterjee, G.; Tembhare, P.; Prasad, M.; Roy Moulik, N.; et al. Machine learning derived genomics driven prognostication for acute myeloid leukemia with RUNX1-RUNX1T1. Leuk. Lymphoma 2020, 61, 3154–3160.
- Patkar, N.; Shaikh, A.F.; Kakirde, C.; Nathany, S.; Ramesh, H.; Bhanshe, P.; Joshi, S.; Chaudhary, S.; Kannan, S.; Khizer, S.H.; et al. A novel machine-learning-derived genetic score correlates with measurable residual disease and is highly predictive of outcome in acute myeloid leukemia with mutated NPM1. Blood Cancer J. 2019, 9, 79.
- Bullinger, L.; Dohner, K.; Bair, E.; Frohling, S.; Schlenk, R.F.; Tibshirani, R.; Dohner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclones in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616.
- Li, Z.; Herold, T.; He, C.; Valk, P.; Chen, P.; Jurinovic, V.; Mansmann, U.; Radmacher, M.; Maharry, K.; Sun, M.; et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloiud leukemia: An international collaborative study. J. Clin. Oncol. 2013, 31, 1172–1181.
- Marcucci, G.; Yan, P.; Maharry, K.; Frankhouser, D.; Nicolet, D.E.; Metzler, K.H.; Kohlschmidt, J.; Mrozek, K.; Wu, Y.Z.; Bussi, D.; et al. Epigenetics meets genetics in acute myeloid leukemia: Clinical impact of a novel seven-gene score. J. Clin. Oncol. 2013, 32, 548–556.
- Ng, S.; Mitchell, A.; Kennedy, J.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukemia. Nature 2016, 540, 433–437.
- Warnat-Herresthal, S.; Perrakis, K.; Taschler, B.; Becker, M.; Babler, K.; Beyer, M.; Gunther, P.; Schulte-Schrepping, J.; Seep, L.; Klee, K.; et al. Scalable prediction of acute myeloid leukemia using high-dimensional machine learning and blood transcriptomics. iScience 2020, 23, 100780.
- Wagner, S.; Vadakekolathu, J.; Taisan, S.K.; Altmann, H.; Bornhauser, M.; Pockley, A.G.; Ball, G.R.; Rutella, S. A parsimonius 3-gene signature predicts clinical outcomes in an acute myeloid leukemia multicohort study. Blood Adv. 2019, 3, 1330–1340.
- Rouphail, Y.; Radakovich, N.; Shreve, J.; Mukherejee, S.; Jha, B.K.; Maciejewski, J.P.; Sekeres, M.A.; Nazha, A. Personalized transcriptomic analyses identify unique signatures that correlate with genomic subtypes in acute myeloid leukemia (AML) using explainable artificial intelligence. Blood 2020, 136 (Suppl. S1), 33–34.
- Gandelman, J.S.; Byrne, M.T.; Mistry, A.M.; Polikowsky, H.G.; Diggins, K.E.; Chen, H.; Lee, S.J.; Arora, M.; Cutler, C.; Flowers, M.; et al. Machine learning reveals chronic graft-versus host disease phenotypes and stratifies survival after stem cell transplant for hematologic malignancies. Haematologica 2019, 104, 189–196.
- Choi, E.J.; Lee, J.H.; Park, J.H.; Park, H.S.; Lee, J.H.; Lee, Y.; Kang, Y.A.; Jeon, M.; Woo, J.M.; Kang, H.; et al. Machine learning-based approach to predict survival after allogeneic hematopoietic cell transplantation in hematologic malignancies. Blood 2020, 136 (Suppl. S1), 33–34.
- Nazha, A.; Hu, Z.H.; Wang, T.; Lindsley, R.C.; Abdel-Azim, H.; Aljurf, M.; Bacher, U.; Bashley, A.; Cahn, J.Y.; Cerny, J.; et al. A personalized prediction model for outcomes after allogeneic hematopoietic cell transplant in patients with myelodysplastic syndromes. Biol. Blood Marrow Transplant. 2020, 26, 2139–2146.
- Lee, S.; Celik, S.; Logsdon, B.A.; Lundberg, S.M.; Martins, T.J.; Oehler, V.G.; Estey, E.H.; Miller, C.P.; Chien, S.; Dai, J.; et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat. Commun. 2018, 9, 42.
- Chen, X.; Chen, H.Y.; Chen, Z.D.; Gong, J.N.; Chen, C.Y.N. A novel artificial intelligence protocol for finding potential inhibitors of acute myeloid leukemia. J. Mater. Chem. B Mater Biol. Med. 2020, 8, 2063–2081.
- Cutler, G.; Fridman, J.S. A machine-learning analysis suggests that FLX925, a FLT3/CDK4/6 kinase inhibitor, is potent against FLT3-wild type tumors via its CDK4/6 activity. Blood 2016, 128, 3520.
- Ko, B.S.; Wang, Y.F.; Li, J.L.; Weng, P.F.; Hsu, S.C.; Hou, H.A.; Huang, H.H.; Yao, M.; Lin, C.T.; Liu, J.H.; et al. Clinically validated machine learning algorithm for detecting residual diseases with multicolor flow cytometry analysis in acute myeloid leukemia and myelodysplastic syndrome. EBioMedicine 2018, 37, 91–100.
- Haferlach, T.; Schmidts, I. The power and potential of integrated diagnostics in acute myeloid leukemia. Br. J. Haematol. 2020, 188, 36–48.
- Letai, A. Functional precision cancer medicine -moving beyond pure genomic. Nat. Med. 2017, 23, 1028–1035.
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531.
- Kurtz, S.E.; Eide, C.A.; Kaempf, A.; Khanna, V.; Savage, S.L.; Rofelty, A.; English, I.; Ho, H.; Pandya, R.; Bolosky, W.J.; et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid- and lymphoid- derived hematologic malignancies. Proc. Natl. Acad. Sci. USA 2017, 114, E7554–E7563.
- Lin, L.; Tong, Y.; Straube, J.; Zhao, J.; Gao, Y.; Bai, P.; Li, J.; Wang, J.; Wang, H.; Wang, X.; et al. Ex-vivo drug testing predicts chemosensitivity in acute myeloid leukemia. J. Leukoc. Biol. 2020, 107, 859–870.
- Erkers, T.; Seashore-Ludlow, B.; Struyf, N.; Marabita, F.; James, T.; Malani, D.; Vesterlund, M.; Pawitan, Y.; Lehmann, S.; Ostling, P.; et al. High-throughput functional ex-vivo drug testing and multi-omics profiling in patients with acute myeloid leukemia. Blood 2019, 134 (Suppl. S1), 4641.
- Martinez-Cuadròn, D.; Gil, C.; Serrano, J.; Rodriguez, G.; Perez-Oteyza, J.; Garcia-Boyero, R.; Jimenez-Bravo, S.; Vives, S.; Vidriales, M.B.; Lavilla, E.; et al. A precision medicine test predicts clinical response after idarubicin and cytarabine induction therapy in AML patients. Leukemia Res. 2019, 76, 1–10.
- Simon, L.; Lavallée, V.P.; Bordeleau, M.E.; Krosl, J.; Baccelli, I.; Boucher, G.; Lenhertz, B.; Chagraoul, J.; MacRae, T.; Ruel, R.; et al. Chemogenomic landscape of RUNX1-mutated AML reveals importance of RUNX1 allele dosage in genetics and glucocorticoid sensitivity. Clin. Cancer Res. 2017, 23, 6969–6983.
- Moison, C.; Lavallée, J.P.; Thiollier, C.; Spinella, J.F.; Boivin, I.; Lemiux, S.; Marinier, A.; Hébert, J.; Savageau, G. Chemogenomic profiling of complex karyotype AML reveals a novel susceptibility to G2/M checkpoint inhibition mediated by HMGA2 overexpression. Blood 2018, 132 (Suppl. S1), 3925.
- Zhang, H.; Wilmot, B.; Bottomly, D.; Kurtz, S.E.; Eide, C.A.; Damnernsawad, A.; Romine, K.; Patel, S.; Druker, B.J.; Mcweeney, S.K.; et al. Biomarkers predicting venetoclax sensitivity and strategies for venetoclax combination treatment. Blood 2018, 132 (Suppl. S1), 175.
- Zhang, H.; Nakauchi, Y.; Kohnkie, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majketi, R.; Tyner, J.W. Integrated analysis of patients samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839.
- Stevens, B.M.; Jones, C.L.; Winters, A.; Gugan, J.; Abbott, D.; Savona, M.R.; Fesik, S.W.; Pollyea, D.A.; Jordan, C.T. PTPN11 mutations confer unique metabolic properties and increase resistance to venetoclax and azacytidine in acute myeloid leukemia. Blood 2018, 132 (Suppl. S1), 909.
- Alfayez, M.; Issa, G.C.; Patel, K.P.; Wang, F.; Wang, X.; Short, N.J.; Cortes, J.E.; Kadia, T.; Ravandi, F.; Pierce, S.; et al. The clinical impact of PTPN11 mutations in adults with acute myeloid leukemia. Leukemia 2020, in press.
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020, 10, 536–551.
- Spinner, M.A.; Alishin, A.; Santaguida, M.T.; Schaffert, S.A.; Zehnder, J.L.; Patterson, A.S.; Gekas, C.; Heiser, D.; Greenberg, P.L. Wx vivo drug screening defines novel drug sensitivity patterns informing personalized therapy in myeloid neoplasms. Blood Adv. 2020, 4, 2768–2778.
- Snijder, B.; Vladimer, G.I.; Krall, N.; Miura, K.; Schmolke, A.S.; Kornauth, C.; de la Fuente, I.O.L.; Choi, H.S.; van der Kouwe, E.; Gultekin, S.; et al. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: Interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017, 4, e595–e606.
- Collignon, A.; Hospital, M.A.; Montersino, C.; Courtier, F.; Charbonnier, A.; Saillard, C.; D’Incan, E.; Mohty, B.; Guille, A.; Adelaide, J.; et al. A chemogenomic approach to identify personalized therapy for patients with relapse or refractory acute myeloid leukemia: Results of a prospective feasibility study. Blood Cancer J. 2020, 10, 64.