Gene therapy has been used as a potential approach to address the diagnosis and treatment of genetic diseases and inherited disorders. In this line, non-viral systems have been exploited as promising alternatives for delivering therapeutic transgenes and proteins. In this entry, we explored how biological barriers are effectively overcome by non-viral systems, usually nanoparticles, to reach an efficient delivery of cargoes.
- gene therapy
- internalization
- nanovehicles
- delivery
1. Introduction
Gene therapy has been considered a promising therapeutic strategy, and it is based on the delivery of genes to treat several acute acquired and inherited diseases [1]. Some examples include, among others, the autosomal or X-linked recessive single-gene disorders (i.e., cystic fibrosis), Severe Combined Immunodeficiency Defect (SCID), emphysema, retinitis pigmentosa, sickle-cell anemia, phenylketonuria, hemophilia, Duchenne Muscular Dystrophy (D.M.D), some autosomal dominant disorders, even polygenic disorders, various forms of cancers, vascular disease, neurodegenerative disorders, inflammatory conditions [2].
Viruses were the first carriers for delivering therapeutic genes, assuring protection, and taking advantage of the virus-life cycle [1]. This type of carrier, known as a viral vector, is one of the most widely used vectors in gene therapy due to its ability to carry genes efficiently and ensure long-term expression [2]. However, these vectors have several disadvantages, such as the potential risk of harmful immune responses [3], the high cost and difficulty related to their preparation [1,4], and the limited size of the genetic sequences that can be inserted into human cells [1,5]. Accordingly, there is a need to look for safer and cheaper alternatives [1]. Hence, non-viral approaches have risen to deal with the limitations of viral systems. Research in this field has attracted significant attention because of the advantages that non-viral systems offer over the viral ones regarding safety, relatively low immune response, and ease of preparation to enable large amounts at low cost [6].
Viruses were the first carriers for delivering therapeutic genes, assuring protection, and taking advantage of the virus-life cycle [1]. This type of carrier, known as a viral vector, is one of the most widely used vectors in gene therapy due to its ability to carry genes efficiently and ensure long-term expression [2]. However, these vectors have several disadvantages, such as the potential risk of harmful immune responses [3], the high cost and difficulty related to their preparation [1][4], and the limited size of the genetic sequences that can be inserted into human cells [1][5]. Accordingly, there is a need to look for safer and cheaper alternatives [1]. Hence, non-viral approaches have risen to deal with the limitations of viral systems. Research in this field has attracted significant attention because of the advantages that non-viral systems offer over the viral ones regarding safety, relatively low immune response, and ease of preparation to enable large amounts at low cost [6].
Non-viral DNA delivery systems are classified into two groups: physical approaches and chemically constructed vectors. Physical approaches rely on a physical force to weaken the cell membrane, thereby facilitating the gene’s insertion into the nucleus [6]. Some strategies following this approach include electroporation, gene gun, ultrasound, and hydrodynamic delivery. Meanwhile, chemically constructed vectors can be prepared by the electrostatic interaction between polycationic derivatives, either lipids or polymers and the anionic phosphate of DNA to form a particle [6]. This complex is known as a polyplex when the interaction occurs between the polymer and DNA or a lipoplex when the DNA interacts with a phospholipid. Moreover, it is possible to chemically build DNA vectors by encapsulation or adsorption within biodegradable spherical structures to yield micro and nanoparticles [1,7]. Other chemically constructed vectors include the conjugation of bioactive compounds such as proteins and peptides in the surface of metal, magnetic, lipid, polymer, and carbon-based nanomaterials [8].
Non-viral DNA delivery systems are classified into two groups: physical approaches and chemically constructed vectors. Physical approaches rely on a physical force to weaken the cell membrane, thereby facilitating the gene’s insertion into the nucleus [6]. Some strategies following this approach include electroporation, gene gun, ultrasound, and hydrodynamic delivery. Meanwhile, chemically constructed vectors can be prepared by the electrostatic interaction between polycationic derivatives, either lipids or polymers and the anionic phosphate of DNA to form a particle [6]. This complex is known as a polyplex when the interaction occurs between the polymer and DNA or a lipoplex when the DNA interacts with a phospholipid. Moreover, it is possible to chemically build DNA vectors by encapsulation or adsorption within biodegradable spherical structures to yield micro and nanoparticles [1][7]. Other chemically constructed vectors include the conjugation of bioactive compounds such as proteins and peptides in the surface of metal, magnetic, lipid, polymer, and carbon-based nanomaterials [8].
DNA plays a crucial role in storing genetic information, which is transcribed into messenger RNA. This transcript serves as the bridge between the genetic information encoded in DNA and its protein translation. Other types of nucleic acids that can potentially control protein expression include interference RNA (RNAi) and antisense oligonucleotides. These therapeutic approaches are similar as they act as a gene silencing mechanism. The ultimate goal of gene therapy is to deliver a transgene into the nucleus (for DNA delivery) or the cytoplasm (e.g., to deliver RNAi) to finally express the therapeutic protein [9]. Therefore, DNA must be complexed with the delivery system or vector that carries the therapeutic gene into the targeted cell, thereby avoiding its degradation and ensuring its final transcription [10].
2. Nanocarriers for the Delivery of Nucleic Acids and Proteins
2.1. Lipid-Based Nanocarriers
Lipid-based nanocarriers include liposomes, lipid nanoparticles (LNPs), and emulsions. These are the most widely used non-viral vectors for nucleic acid (NA) therapy [90]. In the 1980s, phospholipids containing liposomes were first tested to deliver SV40 DNA to monkey kidney cells [91]. Such nanocarriers consist of spherical, self-assembled closed structures with one or several concentric lipid bilayers encircled around an inner aqueous phase (
Lipid-based nanocarriers include liposomes, lipid nanoparticles (LNPs), and emulsions. These are the most widely used non-viral vectors for nucleic acid (NA) therapy [11]. In the 1980s, phospholipids containing liposomes were first tested to deliver SV40 DNA to monkey kidney cells [12]. Such nanocarriers consist of spherical, self-assembled closed structures with one or several concentric lipid bilayers encircled around an inner aqueous phase (
Figure 3A). The lipid coat may be composed of both cationic and ionizable lipids. In vitro and in vivo delivery of DNA, siRNA and mRNA has been enabled by liposomes synthesized from classical cationic lipids such as
1A). The lipid coat may be composed of both cationic and ionizable lipids. In vitro and in vivo delivery of DNA, siRNA and mRNA has been enabled by liposomes synthesized from classical cationic lipids such as
N
-[1-(2, 3-dioleoyloxy)propyl]-
N
,
N
,
N-trimethylammonium chloride (DOTMA), 1,2-dioleoyloxy-3-trimethylammonium propane chloride (DOTAP), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) [90]. Recently, these liposomes demonstrated promising results in delivering mRNA to dendritic cells for cancer immunotherapy [90]. Alternatively, liposomes with ionizable lipids composed of hydrocarbon chains, linkers, and headgroups, are neutral under a physiological pH environment. However, they are ionized and protonated under the acidic conditions of endosomes and lysosomes, which further triggers osmotic lysosomes and endosome rupture [90]. As a result, this type of liposome has been successfully tested for RNAi therapies for hereditary transthyretin-mediated amyloidosis (hATTR), where they demonstrated significant endosome/lysosome escape ability [90]. Some examples of ionizable lipids previously investigated preclinically and clinically are listed in
-trimethylammonium chloride (DOTMA), 1,2-dioleoyloxy-3-trimethylammonium propane chloride (DOTAP), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) [11]. Recently, these liposomes demonstrated promising results in delivering mRNA to dendritic cells for cancer immunotherapy [11]. Alternatively, liposomes with ionizable lipids composed of hydrocarbon chains, linkers, and headgroups, are neutral under a physiological pH environment. However, they are ionized and protonated under the acidic conditions of endosomes and lysosomes, which further triggers osmotic lysosomes and endosome rupture [11]. As a result, this type of liposome has been successfully tested for RNAi therapies for hereditary transthyretin-mediated amyloidosis (hATTR), where they demonstrated significant endosome/lysosome escape ability [11]. Some examples of ionizable lipids previously investigated preclinically and clinically are listed in
.
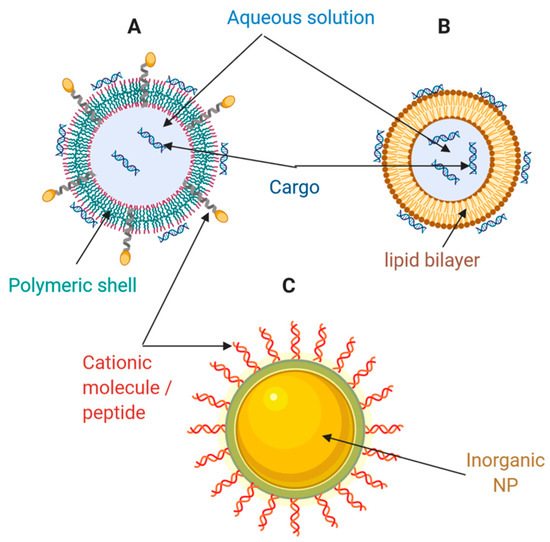
Schematic of different types of nanocarriers for drug and gene delivery. (
) Polymeric nanoparticle; (
) Liposome; (
) Inorganic nanoparticle. Adapted from Weng et al. [11], Molecular Therapy Nucleic Acids, 2020.
The resulting liposome/NA complex is obtained by electrostatic interactions between lipids (positively charged) and NAs (negatively charged). Mainly, nano-complexes derived from the interaction of liposomes and siRNAs are obtained with a slightly positive charge, thereby facilitating interaction with negatively charged cell surfaces [92]. Subsequently, these nano-complexes are delivered to cells for uptake, internalization, escape, release, and expression [90]. Despite this process’s efficiency and simplicity, significant difficulties to overcome include the cationic lipid-associated cytotoxicity, off-target effects, and limited cell types for transfection [90]. Meanwhile, recently developed advanced technologies such as bio-orthogonal liposome fusion, click chemistry, and surface engineering may be combined with lipid nanocarriers for the development of NA delivery systems that are produced more straightforwardly (i.e., with fewer manipulation steps), are more efficient, and exhibit higher precision in cell transfection [93,94,95]. These strategies look for packing and delivering NAs to cells using rapid artificial surface labeling and targeting. Instead of relying on nonspecific electrostatic interactions between the nucleic acid complex and the cell, these methods produce complexes with a bio-orthogonal functional group displayed superficially for adhesion and delivery. For example, liposomes containing ketone groups are synthesized and added to cell culture to enable ketone display on the cell surface. Then, a complementary oxyamine liposome is generated to complex with nucleic acids. Later, the oxyamine/nucleic acid lipoplex is added to the ketone from cells. Finally, oxime formation occurs at the cell surface, and the nucleic acid is endocytosed and release within the cell [93].
The resulting liposome/NA complex is obtained by electrostatic interactions between lipids (positively charged) and NAs (negatively charged). Mainly, nano-complexes derived from the interaction of liposomes and siRNAs are obtained with a slightly positive charge, thereby facilitating interaction with negatively charged cell surfaces [13]. Subsequently, these nano-complexes are delivered to cells for uptake, internalization, escape, release, and expression [11]. Despite this process’s efficiency and simplicity, significant difficulties to overcome include the cationic lipid-associated cytotoxicity, off-target effects, and limited cell types for transfection [11]. Meanwhile, recently developed advanced technologies such as bio-orthogonal liposome fusion, click chemistry, and surface engineering may be combined with lipid nanocarriers for the development of NA delivery systems that are produced more straightforwardly (i.e., with fewer manipulation steps), are more efficient, and exhibit higher precision in cell transfection [14][15][16]. These strategies look for packing and delivering NAs to cells using rapid artificial surface labeling and targeting. Instead of relying on nonspecific electrostatic interactions between the nucleic acid complex and the cell, these methods produce complexes with a bio-orthogonal functional group displayed superficially for adhesion and delivery. For example, liposomes containing ketone groups are synthesized and added to cell culture to enable ketone display on the cell surface. Then, a complementary oxyamine liposome is generated to complex with nucleic acids. Later, the oxyamine/nucleic acid lipoplex is added to the ketone from cells. Finally, oxime formation occurs at the cell surface, and the nucleic acid is endocytosed and release within the cell [14].
The ability of lipid NPs to internalize and escape endosomes has been facilitated by membrane fusion events [96] (
The ability of lipid NPs to internalize and escape endosomes has been facilitated by membrane fusion events [17] (
Figure 4). In general, through hydrophobic interactions, the liposomal envelope fuses with the endosomal membrane [73]. These interactions are facilitated by the protonation of anionic groups of the liposomal envelope, thereby allowing the release of encapsulated cargoes into the cytosol [97]. Also, by incorporating cholesterol within liposomal structures, it has been possible to increase the contact sites needed for lipid mixing and pore fusion expansion [98].
2). In general, through hydrophobic interactions, the liposomal envelope fuses with the endosomal membrane [18]. These interactions are facilitated by the protonation of anionic groups of the liposomal envelope, thereby allowing the release of encapsulated cargoes into the cytosol [19]. Also, by incorporating cholesterol within liposomal structures, it has been possible to increase the contact sites needed for lipid mixing and pore fusion expansion [20].
Some ionizable lipids employed in the production of liposomes for gene silencing.
| Abbreviation | Chemical Name | Findings/Relevant Data | Reference |
|---|---|---|---|
| DLin-MC3-DMA | 6Z,9Z,28Z,31Z-heptatriaconta-6,9,28,31-tetraen-19-yl-4-(dimethylamino)-butanoate | Used for the first time in Patisiran (liposome formulation). | [90][11] |
| DLin-KC2-DMA | 1,2-dilinoleyl-4-(2-dimethylaminoethyl)-1,3-dioxolane | Demonstrated to have in vivo activity at siRNA doses as low as 0.01 mg/kg in rodents and 0.1 mg/kg in nonhuman primates. | [99][21] |
| L319 | di((Z)-non-2-en-1-yl)-9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate)-9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate | Biodegradable lipid displaying rapid elimination from plasma and tissues, substantially improved tolerability in preclinical studies. | [100][22] |
| C12-200 | - | Over 95% silencing at a dose of 0.03 mg/kg in non-human primates and 0.01 mg/kg in mice. | [101][23] |
| cKK-E12 | - | Over 95% silencing at a dose of 0.3 mg/kg in nonhuman primates. Toxicity studies showed that cKK-E12 was well tolerated in rats at a dose of 1 mg/kg. | [102][24] |
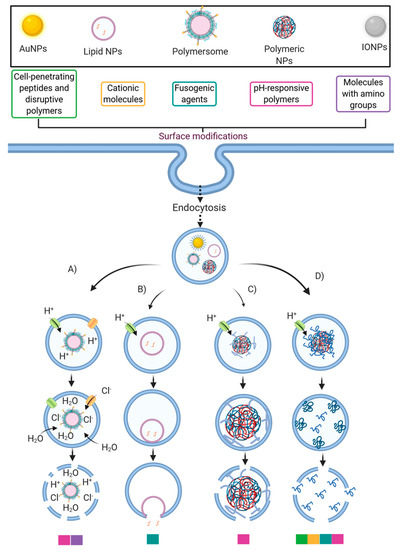
Strategies to improve the endosomal escape ability of some nanocarriers. This schematic illustrates the association between the more typically employed surface modifications and the endosomal escape mechanisms (EEMs): (
) Proton-Sponge and osmotic lysis; (
) Membrane fusion; (
) Particle swelling, and (
) Membrane translocation and destabilization. The color legend below the EEMs corresponds to the surface modifications that likely trigger the corresponding EEM. Created with
.
2.2. Polymeric-Based Nanocarriers
Polymers and their derivatives are an attractive alternative for the development of drug delivery systems. In this regard, polysomes for NA delivery can be formed with an ample variety of materials, including NPs, nano-micelles, dendrimers, hydrogels, and nanoemulsions (
Figure 3B) [90]. Some of these developments have even reached clinical stages, and for instance, the natural polymer cyclodextrin enabled the first siRNAs delivery application at the clinical level [11]. Cyclodextrin is a natural macrocyclic oligosaccharide with internal hydrophobic and external hydrophilic structures, which may interact with NAs externally and improve their stability. Nonetheless, a major challenge for a full translation into the clinic is its relatively high toxicity [103,104,105]. Besides cyclodextrin, other natural polymers, including chitosan [106,107], hyaluronic acid [108], dextran [109], and gelatin [110], have been widely studied as promising candidates for NA delivery systems.
1B) [11]. Some of these developments have even reached clinical stages, and for instance, the natural polymer cyclodextrin enabled the first siRNAs delivery application at the clinical level [25]. Cyclodextrin is a natural macrocyclic oligosaccharide with internal hydrophobic and external hydrophilic structures, which may interact with NAs externally and improve their stability. Nonetheless, a major challenge for a full translation into the clinic is its relatively high toxicity [26][27][28]. Besides cyclodextrin, other natural polymers, including chitosan [29][30], hyaluronic acid [31], dextran [32], and gelatin [33], have been widely studied as promising candidates for NA delivery systems.
Table 2 shows some synthetic polymers employed in NA delivery. PEI (polyethyleneimine) has been demonstrated to be an ideal cationic delivery carrier for NAs [90,111]. Interestingly, block copolymers of PEG-PEI instead of PEI alone have been considered excellent alternatives to decreasing toxicity and improving its performance, mainly due to the ability of PEG to prevent opsonization and avoid specific interaction with blood cells [112]. However, it is important to remark that this property is a function of PEG density and particle size in nanocarriers, as discussed above and demonstrated by Walkey et al. [57] and Han et al. [113]. One of the most important pH-sensitive cationic polymers, pDMAEMA, is widely used for DNA, siRNA, mRNA, and miRNA delivery with acceptable cytotoxicity and combined transfection efficiency [90,114,115,116,117]. Surprisingly, besides serving as delivery vehicles, some polymers also exhibit therapeutic properties [90]. For example, a polymer derivative of the drug metformin, with anti-cancer and anti-diabetic effects, was found to have the ability to deliver siRNA for RNAi therapy without losing its anti-cancer properties [118].
shows some synthetic polymers employed in NA delivery. PEI (polyethyleneimine) has been demonstrated to be an ideal cationic delivery carrier for NAs [11][34]. Interestingly, block copolymers of PEG-PEI instead of PEI alone have been considered excellent alternatives to decreasing toxicity and improving its performance, mainly due to the ability of PEG to prevent opsonization and avoid specific interaction with blood cells [35]. However, it is important to remark that this property is a function of PEG density and particle size in nanocarriers, as discussed above and demonstrated by Walkey et al. [36] and Han et al. [37]. One of the most important pH-sensitive cationic polymers, pDMAEMA, is widely used for DNA, siRNA, mRNA, and miRNA delivery with acceptable cytotoxicity and combined transfection efficiency [11][38][39][40][41]. Surprisingly, besides serving as delivery vehicles, some polymers also exhibit therapeutic properties [11]. For example, a polymer derivative of the drug metformin, with anti-cancer and anti-diabetic effects, was found to have the ability to deliver siRNA for RNAi therapy without losing its anti-cancer properties [42].
On the one hand, the anti-cancer property of metformin is mainly attributed to the activation of AMP-activated protein kinase (AMPK) [119,120] and inhibition of the mammalian target of rapamycin (mTOR) [121,122]. On the other hand, this polymer’s ability to deliver siRNA may be due to the presence of guanidine groups in its structure. The guanidine has been found to pass through the non-polar membrane of a cell and even across tissue barriers by possibly forming a bidentate hydrogen bond with anionic cell surface phosphate, carboxylates, and/or sulfates on the cell surface [123,124]. As another example, a near-infrared absorbing, dendronized, and semiconducting polymer delivered DNA efficiently and controlled gene expression spatiotemporally together with a heat-inducible promoter [125].
On the one hand, the anti-cancer property of metformin is mainly attributed to the activation of AMP-activated protein kinase (AMPK) [43][44] and inhibition of the mammalian target of rapamycin (mTOR) [45][46]. On the other hand, this polymer’s ability to deliver siRNA may be due to the presence of guanidine groups in its structure. The guanidine has been found to pass through the non-polar membrane of a cell and even across tissue barriers by possibly forming a bidentate hydrogen bond with anionic cell surface phosphate, carboxylates, and/or sulfates on the cell surface [47][48]. As another example, a near-infrared absorbing, dendronized, and semiconducting polymer delivered DNA efficiently and controlled gene expression spatiotemporally together with a heat-inducible promoter [49].
Some synthetic polymers frequently used for nucleic acids (NA) delivery.
| Abbreviation | Chemical Name | Findings/Relevant Properties | Reference |
|---|---|---|---|
| PEI | Polyethylenimine | The most widely used. It is the organic macromolecule with the highest cationic-charge-density potential. | [90,111][11][34] |
| pDMAEMA | Poly(2-dimethylamino)ethyl methacrylate | Extensively studied and widely used for the delivery of DNA, siRNA, mRNA and miRNA. It had tertiary amines in its structure. | [126][50] |
| hDD90-118 | - | An hyperbranched poly(beta amino ester) capable of save and effective delivering of mRNA to lung epithelium. | [127][51] |
| N5 | - | An assembly of poly A binding proteins and cationic polypeptides for enhanced mRNA delivery. | [128][52] |
| PAA8k-(2-3-2) | - | A poly(acrylic acid) scaffold grafted with oligoalkylamines promoting enhanced mRNA delivery. | [129][53] |
Other types of polymersomes, such as dendrimers, and polymeric micelles, have proven to be valuable nanocarriers. Dendrimers are highly branched functional polymer-based nanocarriers that comprise an inner core, an amidoamine backbone, and multiple terminal amine groups. Due to this conformation, dendrimers feature plenty of compartment space for loading NAs [90,130,131]. Notably, the coupling of the G0-C14 dendrimer with PEG-PLA polymer leads to cationic amphiphilic dendrimers that have exhibited synergistic anti-cancer effects through the encapsulation of both chemotherapeutic drugs in their hydrophobic interlayer and NAs in their hydrophilic cavity [90,132]. Polymeric micelles, which have received considerable attention in polymer chemistry, are self-assembled from synthetic block copolymers or graft copolymers with an inner hydrophobic core and an outer hydrophilic shell [100]. NAs are more favorably incorporated into micelles’ inner core formed from positively charged polymers through ionic interactions. This enables the incorporation of NAs, such as CRISPR-Cas9 and siRNAs, into polymeric micelles with high stability [133,134]. Davis et al. [135] developed a cyclodextrin-containing polymer to conjugate camptothecin (CPT) with near-neutral zeta potential that properly self-assembles into nanoparticles of about 30 nm diameter. The nanoparticles enter the tumor cells and slowly release the CPT, causing them to disassemble into individual polymer chains that are sufficiently small to be cleared renally. Additionally, the nanoparticles showed long circulation half-lives in animals and humans and targeted localization in tumors. These encouraging results suggest that polymeric micelles can be promising nanocarriers for in vivo and in vitro NA delivery.
Other types of polymersomes, such as dendrimers, and polymeric micelles, have proven to be valuable nanocarriers. Dendrimers are highly branched functional polymer-based nanocarriers that comprise an inner core, an amidoamine backbone, and multiple terminal amine groups. Due to this conformation, dendrimers feature plenty of compartment space for loading NAs [11][54][55]. Notably, the coupling of the G0-C14 dendrimer with PEG-PLA polymer leads to cationic amphiphilic dendrimers that have exhibited synergistic anti-cancer effects through the encapsulation of both chemotherapeutic drugs in their hydrophobic interlayer and NAs in their hydrophilic cavity [11][56]. Polymeric micelles, which have received considerable attention in polymer chemistry, are self-assembled from synthetic block copolymers or graft copolymers with an inner hydrophobic core and an outer hydrophilic shell [22]. NAs are more favorably incorporated into micelles’ inner core formed from positively charged polymers through ionic interactions. This enables the incorporation of NAs, such as CRISPR-Cas9 and siRNAs, into polymeric micelles with high stability [57][58]. Davis et al. [59] developed a cyclodextrin-containing polymer to conjugate camptothecin (CPT) with near-neutral zeta potential that properly self-assembles into nanoparticles of about 30 nm diameter. The nanoparticles enter the tumor cells and slowly release the CPT, causing them to disassemble into individual polymer chains that are sufficiently small to be cleared renally. Additionally, the nanoparticles showed long circulation half-lives in animals and humans and targeted localization in tumors. These encouraging results suggest that polymeric micelles can be promising nanocarriers for in vivo and in vitro NA delivery.
Various strategies have been proposed to facilitate endosomal escape through different pathways. For example, cationic polymers (such as PEI) and others with pendant amine groups exhibit a buffering capacity that enables escape from endosomal entrapment through the proton-sponge mechanism and the osmotic lysis effect [96]. By this pathway, protons are pumped by an ATPase into the endosome during its acidification to reach the desired pH to start their maturation. However, amine groups’ presence may produce a buffer effect and sequester the incoming protons because pKa values of such groups are in the range of endolysosomal pH values [136]. Consequently, they can maintain a constant pH, altering the Nernst equilibrium potential. This results in an influx of chloride counterions and water molecules to restore such an equilibrium, thereby producing a pressure increase, which eventually disrupts the endosomal membrane [137] (
Various strategies have been proposed to facilitate endosomal escape through different pathways. For example, cationic polymers (such as PEI) and others with pendant amine groups exhibit a buffering capacity that enables escape from endosomal entrapment through the proton-sponge mechanism and the osmotic lysis effect [17]. By this pathway, protons are pumped by an ATPase into the endosome during its acidification to reach the desired pH to start their maturation. However, amine groups’ presence may produce a buffer effect and sequester the incoming protons because pKa values of such groups are in the range of endolysosomal pH values [60]. Consequently, they can maintain a constant pH, altering the Nernst equilibrium potential. This results in an influx of chloride counterions and water molecules to restore such an equilibrium, thereby producing a pressure increase, which eventually disrupts the endosomal membrane [61] (
Figure 4). Another endosomal escape mechanism that pH-responsive polymeric NPs may follow is through particle swelling [138]. As the pH lowers during the endo/lysosome’s maturation process, polymeric NPs tend to swell within the endosomal vesicle. Finally, the endosome’s lysis is reached by either the exerted mechanical strain during swelling or the proton-sponge effect [96,138] (
2). Another endosomal escape mechanism that pH-responsive polymeric NPs may follow is through particle swelling [62]. As the pH lowers during the endo/lysosome’s maturation process, polymeric NPs tend to swell within the endosomal vesicle. Finally, the endosome’s lysis is reached by either the exerted mechanical strain during swelling or the proton-sponge effect [17][62] (
Figure 4).
2).
2.3. Inorganic Nanomaterials
Inorganic NPs have become an attractive therapeutic NA and drug delivery approach mainly because they feature several advantages: precise size control, tunable surface properties, and high drug loading efficiency (
2.4. Cell-Penetrating Peptides
Cell-penetrating peptides (CPPs) are attractive nanocarriers for the delivery of nucleic acids and proteins. The first application of CPPs was in the delivery of nucleic acids to cells through electrostatic interactions [222]. In general, the delivery of nucleic acids through this approach offers numerous advantages such as protection of cargoes from degradation, effective internalization into specific target cells, improved release of cargoes intracellularly either at the cytoplasmic level (e.g., antisense nucleotides, RNAi therapies) or the nucleus (e.g., plasmid DNA), high biological activity at low doses, negligible cytotoxicity, and good biosafety for therapeutic studies in vivo [223].
Cell-penetrating peptides (CPPs) are attractive nanocarriers for the delivery of nucleic acids and proteins. The first application of CPPs was in the delivery of nucleic acids to cells through electrostatic interactions [63]. In general, the delivery of nucleic acids through this approach offers numerous advantages such as protection of cargoes from degradation, effective internalization into specific target cells, improved release of cargoes intracellularly either at the cytoplasmic level (e.g., antisense nucleotides, RNAi therapies) or the nucleus (e.g., plasmid DNA), high biological activity at low doses, negligible cytotoxicity, and good biosafety for therapeutic studies in vivo [64].
Several CPP-based conjugates have been synthesized and tested for the delivery of siRNA. For example, Kumar et al. [224] developed siRNA delivery nanosystems for the central nervous system based on a small peptide derived from the rabies virus glycoprotein (RVG, a ligand for acetylcholine receptor) modified with polyarginine (Arg9). In vitro studies showed effective gene silencing and protection against the fatal viral encephalitis in a mouse model. Eguchi et al. [225,226] produced a nanovehicle composed of a TAT fusion protein and a double-stranded RNA-binding domain to efficiently deliver epidermal growth factor receptor (EGFR) and AKT serine/threonine kinase 2 (Akt2) siRNAs to intracranial glioblastoma tumors in a mouse model. In parallel, non-covalent approaches allowed developing stable complexes of CPPs to deliver siRNA. Indeed, the first non-covalent approach enabled the production of complexes with the MPG peptide (derived from the hydrophobic fusion peptide of HIV-1 gp41 plus the hydrophilic NLS of SV40 large T antigen) [227]. These complexes facilitated the delivery of siRNAs targeting OCT-4 into mouse blastocytes and subsequently silencing cyclin B1 (a cell cycle regulator) to reduce cell differentiation and proliferation, respectively [228]. Moreover, an amphipathic CPP named
Several CPP-based conjugates have been synthesized and tested for the delivery of siRNA. For example, Kumar et al. [65] developed siRNA delivery nanosystems for the central nervous system based on a small peptide derived from the rabies virus glycoprotein (RVG, a ligand for acetylcholine receptor) modified with polyarginine (Arg9). In vitro studies showed effective gene silencing and protection against the fatal viral encephalitis in a mouse model. Eguchi et al. [66][67] produced a nanovehicle composed of a TAT fusion protein and a double-stranded RNA-binding domain to efficiently deliver epidermal growth factor receptor (EGFR) and AKT serine/threonine kinase 2 (Akt2) siRNAs to intracranial glioblastoma tumors in a mouse model. In parallel, non-covalent approaches allowed developing stable complexes of CPPs to deliver siRNA. Indeed, the first non-covalent approach enabled the production of complexes with the MPG peptide (derived from the hydrophobic fusion peptide of HIV-1 gp41 plus the hydrophilic NLS of SV40 large T antigen) [68]. These complexes facilitated the delivery of siRNAs targeting OCT-4 into mouse blastocytes and subsequently silencing cyclin B1 (a cell cycle regulator) to reduce cell differentiation and proliferation, respectively [69]. Moreover, an amphipathic CPP named
Cady, containing arginine and tryptophan residues effectively formed stable complexes with siRNA to efficiently achieve gene silencing in both suspension and cell lines such as human osteosarcoma U2OS, THP1 monocytes, human umbilical vein endothelial and mouse 3T3C cells [229].
, containing arginine and tryptophan residues effectively formed stable complexes with siRNA to efficiently achieve gene silencing in both suspension and cell lines such as human osteosarcoma U2OS, THP1 monocytes, human umbilical vein endothelial and mouse 3T3C cells [70].
To significantly improve siRNA delivery systems’ potency, researchers have proposed the stearylation of CPPs. For instance, a stearyl-TP10 analog modified with trifluoromethylquinoline was used to increase endosomal escape and effective siRNA delivery in Jurkat cells and human umbilical vein endothelial cells (HUVEC) [230]. Similarly, the STR-KV peptide (stearyl-HHHKKKVVVVVV) complexed with siRNA targeting the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) exhibited 80–87% gene silencing efficiency and low cytotoxicity [231]. These studies showed that stearylation of CPPs holds a significant promise as a novel alternative to increase the efficiency in siRNA delivery systems.
To significantly improve siRNA delivery systems’ potency, researchers have proposed the stearylation of CPPs. For instance, a stearyl-TP10 analog modified with trifluoromethylquinoline was used to increase endosomal escape and effective siRNA delivery in Jurkat cells and human umbilical vein endothelial cells (HUVEC) [71]. Similarly, the STR-KV peptide (stearyl-HHHKKKVVVVVV) complexed with siRNA targeting the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) exhibited 80–87% gene silencing efficiency and low cytotoxicity [72]. These studies showed that stearylation of CPPs holds a significant promise as a novel alternative to increase the efficiency in siRNA delivery systems.
The use of CPPs also has demonstrated to facilitate the intracellular delivery of diverse proteins and peptides. For example, a system composed of β-galactosidase linked to the TAT peptide exhibited improved blood-brain barrier penetration after intraperitoneal administration [232]. Other advances have shown the effective delivery of anti-apoptotic proteins into cells through their conjugation to CPPs. For instance, Cao et al. [233] obtained protective effects in neurons of a murine middle cerebral artery occlusion model by conjugating the Bcl-xL protein to the TAT CPP. Similarly, a peptide inhibitor of the apoptotic protease-activating factor (Apaf-1) was modified by its conjugation to the CPPs penetratin and Tat. Both CPPs enhanced cellular uptake, but the penetratin conjugate was more effective at inhibiting apoptosis, likely due to the Tat conjugate’s higher cytotoxicity [234]. CPP-mediated delivery of peptides and proteins has mainly been implemented to address cell penetration and targeting to tumors. For example, p53-derived peptides conjugated with the TAT or polyarginine peptides were injected into a peritoneal carcinomatosis mouse model with increased mice survival results [235]. Additionally, a complex composed of a peptide inhibiting casein kinase 2 (P15) activity was injected in mice. to promote enhanced anti-tumor effects [235]. The delivery of proteins and peptides may potentially be improved by their conjugation to CPPs. Consequently, this might provide a green light to developing a more comprehensive variety of nanovehicles concerning the treatment of different types of malignant diseases.
The use of CPPs also has demonstrated to facilitate the intracellular delivery of diverse proteins and peptides. For example, a system composed of β-galactosidase linked to the TAT peptide exhibited improved blood-brain barrier penetration after intraperitoneal administration [73]. Other advances have shown the effective delivery of anti-apoptotic proteins into cells through their conjugation to CPPs. For instance, Cao et al. [74] obtained protective effects in neurons of a murine middle cerebral artery occlusion model by conjugating the Bcl-xL protein to the TAT CPP. Similarly, a peptide inhibitor of the apoptotic protease-activating factor (Apaf-1) was modified by its conjugation to the CPPs penetratin and Tat. Both CPPs enhanced cellular uptake, but the penetratin conjugate was more effective at inhibiting apoptosis, likely due to the Tat conjugate’s higher cytotoxicity [75]. CPP-mediated delivery of peptides and proteins has mainly been implemented to address cell penetration and targeting to tumors. For example, p53-derived peptides conjugated with the TAT or polyarginine peptides were injected into a peritoneal carcinomatosis mouse model with increased mice survival results [76]. Additionally, a complex composed of a peptide inhibiting casein kinase 2 (P15) activity was injected in mice. to promote enhanced anti-tumor effects [76]. The delivery of proteins and peptides may potentially be improved by their conjugation to CPPs. Consequently, this might provide a green light to developing a more comprehensive variety of nanovehicles concerning the treatment of different types of malignant diseases.
References
- Ibraheem, D.; Elaissari, A.; Fessi, H. Gene therapy and DNA delivery systems. Int. J. Pharm. 2014, 459, 70–83.
- Nayerossadat, N.; Ali, P.A.; Maedeh, T. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27.
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722.
- Santos, S.A.; Vidigal, P.M.; Thrimawithana, A.; Betancourth, B.M.; Guimarães, L.M.; Templeton, M.D.; Alfenas, A.C. Comparative genomic and transcriptomic analyses reveal different pathogenicity-related genes among three eucalyptus fungal pathogens. Fungal Genet. Biol. 2020, 137, 103332.
- Nagasaki, T.; Shinkai, S. The concept of molecular machinery is useful for design of stimuli-responsive gene delivery systems in the mammalian cell. J. Incl. Phenom. Macrocycl. Chem. 2007, 58, 205–219.
- Gao, X.; Kim, K.-S.; Liu, D. Nonviral gene delivery: What we know and what is next. AAPS J. 2007, 9, E92–E104.
- Lin, G.; Li, L.; Panwar, N.; Wang, J.; Tjin, S.C.; Wang, X.; Yong, K.T. Non-viral gene therapy using multifunctional nanoparticles: Status, challenges, and opportu-nities. Coord. Chem. Rev. 2018, 374, 133–152.
- Malmsten, M. Inorganic nanomaterials as delivery systems for proteins, peptides, DNA, and siRNA. Curr. Opin. Colloid Interface Sci. 2013, 18, 468–480.
- Botto, C.; Augello, G.; Amore, E.; Emma, M.R.; Azzolina, A.; Cavallaro, G.; Cervello, M.; Bondì, M.L. Cationic Solid Lipid Nanoparticles as Non Viral Vectors for the Inhibition of Hepatocellular Carcinoma Growth by RNA Interference. J. Biomed. Nanotechnol. 2018, 14, 1009–1016.
- Gardlík, R.; Pálffy, R.; Hodosy, J.; Lukács, J.; Turna, J.; Celec, P. Vectors and delivery sys-tems in gene therapy. Med. Sci. Monit. 2005, 11, RA110–RA121.
- Weng, Y.; Huang, Q.; Li, C.; Yang, Y.; Wang, X.; Yu, J.; Huang, Y.; Liang, X.-J. Improved Nucleic Acid Therapy with Advanced Nanoscale Biotechnology. Mol. Ther. Nucleic Acids 2020, 19, 581–601.
- Fraley, R.; Subramani, S.; Berg, P.; Papahadjopoulos, D. Introduction of lipo-some-encapsulated SV40 DNA into cells. J. Biol. Chem. 1980, 255, 10431–10435.
- Ozpolat, B.; Sood, A.K.; Lopez-Berestein, G. Liposomal siRNA nanocarriers for cancer therapy. Adv. Drug Deliv. Rev. 2014, 66, 110–116.
- O’Brien, P.J.; Elahipanah, S.; Rogozhnikov, D.; Yousaf, M.N. Bio-Orthogonal Mediated Nucleic Acid Transfection of Cells via Cell Surface Engineering. ACS Cent. Sci. 2017, 3, 489–500.
- Li, L.; Zahner, D.; Su, Y.; Gruen, C.; Davidson, G.; Levkin, P.A. A biomimetic lipid library for gene delivery through thiol-yne click chemistry. Biomaterials 2012, 33, 8160–8166.
- Li, Y.; Liu, R.; Shi, Y.; Zhang, Z.; Zhang, X. Zwitterionic Poly(carboxybetaine)-based Cationic Liposomes for Effective Delivery of Small Interfering RNA Therapeutics without Accelerated Blood Clearance Phenomenon. Theranostics 2015, 5, 583–596.
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjug. Chem. 2019, 30, 263–272.
- Rueda-Gensini, L.; Cifuentes, J.; Castellanos, M.C.; Puentes, P.R.; Serna, J.A.; Muñoz-Camargo, C.; Cruz, J.C. Tailoring iron oxide nanoparticles for efficient cellular internalization and endoso-mal escape. Nanomaterials 2020, 10, 1816.
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P. Nanoescapology: Progress toward under-standing the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452.
- Pozzi, D.; Marchini, C.; Cardarelli, F.; Amenitsch, H.; Garulli, C.; Bifone, A.; Caracciolo, G. Transfection efficiency boost of cholesterol-containing lipoplexes. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 2335–2343.
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176.
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578.
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869.
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960.
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555.
- Mellet, C.O.; Fernández, J.M.G.; Benito, J.M. Cyclodextrin-based gene delivery systems. Chem. Soc. Rev. 2011, 40, 1586–1608.
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668.
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47.
- Sun, P.; Huang, W.; Jin, M.; Wang, Q.; Fan, B.; Kang, L.; Gao, Z. Chitosan-based nanoparticles for survivin targeted siRNA delivery in breast tumor therapy and preventing its metastasis. Int. J. Nanomed. 2016, 11, 4931–4945.
- Siahmansouri, H.; Somi, M.H.; Babaloo, Z.; Baradaran, B.; Jadidi-Niaragh, F.; Atyabi, F.; Mohammadi, H.; Ahmadi, M.; Yousefi, M. Effects of HMGA2 siRNA and doxorubicin dual delivery by chitosan nanoparticles on cytotoxicity and gene expression of HT-29 colorectal cancer cell line. J. Pharm. Pharmacol. 2016, 68, 1119–1130.
- Park, K.; Yang, J.A.; Lee, M.Y.; Lee, H.; Hahn, S.K. Reducible hyaluronic acid-siRNA conjugate for target specific gene silencing. Bioconjug. Chem. 2013, 24, 1201–1209.
- Kim, J.S.; Oh, M.H.; Park, J.Y.; Park, T.G.; Nam, Y.S. Protein-resistant, reductively dissociable polyplex-es for in vivo systemic delivery and tumor-targeting of siRNA. Biomaterials 2013, 34, 2370–2379.
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Ther. Methods Clin. Dev. 2019, 12, 1–18.
- Boussif, O.; Lezoualc’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301.
- Jiang, D.; Wang, M.; Wang, T.; Zhang, B.; Liu, C.; Zhang, N. Multifunctionalized polyethyleneimine-based nanocarriers for gene and chemotherapeutic drug combination therapy through one-step assembly strategy. Int. J. Nanomed. 2017, 12, 8681–8698.
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147.
- Han, X.; Li, Z.; Sun, J.; Luo, C.; Li, L.; Liu, Y.; Du, Y.; Qiu, S.; Ai, X.; Wu, C.; et al. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin deliv-ery: Probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J. Control. Release 2015, 197, 29–40.
- Lu, H.H.; Huang, C.H.; Shiue, T.Y.; Wang, F.S.; Chang, K.K.; Chen, Y.; Peng, C.H. Erratum: Highly efficient gene release in spatiotemporal precision approached by light and pH dual responsive copolymers. Chem. Sci. 2019, 10, 284–292.
- Cheng, H.; Wu, Z.; Wu, C.; Wang, X.; Liow, S.S.; Li, Z.; Wu, Y.-L. Overcoming STC2 mediated drug resistance through drug and gene co -delivery by PHB-PDMAEMA cationic polyester in liver cancer cells. Mater. Sci. Eng. C 2018, 83, 210–217.
- Ramírez-Acosta, C.M.; Cifuentes, J.; Castellanos, M.C.; Moreno, R.J.; Muñoz-Camargo, C.; Cruz, J.C.; Reyes, L.H. PH-Responsive, Cell-Penetrating, Core/Shell Magnetite/Silver Nanoparticles for the Delivery of Plasmids: Preparation, Characterization, and Preliminary In Vitro Evaluation. Pharmaceutics 2020, 12, 561.
- Ramírez-Acosta, C.M.; Cifuentes, J.; Cruz, J.C.; Reyes, L.H. Patchy Core/Shell, Magnetite/Silver Na-noparticles via Green and Facile Synthesis: Routes to Assure Biocompatibility. Nanomatererials 2020, 10, 1857.
- Zhao, Y.; Wang, W.; Guo, S.; Wang, Y.; Miao, L.; Xiong, Y.; Huang, L. PolyMetformin combines carrier and anticancer activities for in vivo siRNA delivery. Nat. Commun. 2016, 7, 11822.
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156.
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP ki-nase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273.
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin Inhibits Mammalian Target of Rapamycin–Dependent Translation Initiation in Breast Cancer Cells. Cancer Res. 2007, 67, 10804–10812.
- Vázquez-Martín, A.; Oliveras-Ferraros, C.; del Barco, S.; Martín-Castillo, B.; Menéndez, J.A. MTOR in-hibitors and the anti-diabetic biguanide metformin: New insights into the molecular management of breast cancer resistance to the HER2 tyrosine kinase inhibitor lapatinib (Tykerb®). Clin. Transl. Oncol. 2009, 11, 455–459.
- Stanzl, E.G.; Trantow, B.M.; Vargas, J.R.; Wender, P.A. Fifteen years of cell-penetrating, guanidini-um-rich molecular transporters: Basic science, research tools, and clinical applications. Acc. Chem. Res. 2013, 46, 2944–2954.
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702.
- Lyu, Y.; Cui, D.; Sun, H.; Miao, Y.; Duan, H.; Pu, K. Dendronized Semiconducting Polymer as Photothermal Nanocarrier for Remote Activation of Gene Expression. Angew. Chem. Int. Ed. 2017, 56, 9155–9159.
- Olden, B.R.; Cheng, Y.; Yu, J.L.; Pun, S.H. Cationic polymers for non-viral gene delivery to human T cells. J. Control. Release 2018, 282, 140–147.
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31, e1805116.
- Li, J.; He, Y.; Wang, W.; Wu, C.; Hong, C.; Hammond, P.T. Polyamine-Mediated Stoichiometric Assembly of Ribonucleoproteins for Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2017, 56, 13709–13712.
- Jarzębińska, A.; Pasewald, T.; Lambrecht, J.; Mykhaylyk, O.; Kümmerling, L.; Beck, P.; Hasenpusch, G.; Rudolph, C.; Plank, C.; Dohmen, C. A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2016, 55, 9591–9595.
- Cheng, Q.; Wei, T.; Jia, Y.; Farbiak, L.; Zhou, K.; Zhang, S.; Wei, Y.; Zhu, H.; Siegwart, D.J. Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I. Adv. Mater. 2018, 30, e1805308.
- Sharfstein, S.T. Non-protein biologic therapeutics. Curr. Opin. Biotechnol. 2018, 53, 65–75.
- Li, X.; Sun, A.-N.; Liu, Y.-J.; Zhang, W.-J.; Pang, N.; Cheng, S.-X.; Qi, X.-R. Amphiphilic dendrimer engineered nanocarrier systems for co-delivery of siRNA and paclitaxel to matrix metalloproteinase-rich tumors for synergistic therapy. NPG Asia Mater. 2018, 10, 238–254.
- Navarro, G.; Pan, J.; Torchilin, V.P. Micelle-like Nanoparticles as Carriers for DNA and siRNA. Mol. Pharm. 2015, 12, 301–313.
- Lao, Y.H.; Li, M.; Gao, M.A.; Shao, D.; Chi, C.W.; Huang, D.; Chakraborty, S.; Ho, T.C.; Jiang, W.; Wang, H.X.; et al. HPV Oncogene Manipulation Using Nonvirally Delivered CRISPR/Cas9 or Natronobacte-rium gregoryi Argonaute. Adv. Sci. 2018, 5, 1700540.
- Davis, M.E. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptoth-ecin. Adv. Drug Deliv. Rev. 2009, 61, 1189–1192.
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416.
- Vermeulen, L.M.; De Smedt, S.C.; Remaut, K.; Braeckmans, K. The proton sponge hypothesis: Fable or fact? Eur. J. Pharm. Biopharm. 2018, 129, 184–190.
- Hu, Y.; Litwin, T.; Nagaraja, A.R.; Kwong, B.; Katz, J.; Watson, N.; Irvine, D.J. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano Lett. 2007, 7, 3056–3064.
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86.
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258.
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43.
- Eguchi, A.; Dowdy, S.F. Efficient siRNA delivery by novel PTD-DRBD fusion proteins. Cell Cycle 2010, 9, 424–425.
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotechnol. 2009, 27, 567–571.
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene de-livery system MPG: Implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003, 31, 2717–2724.
- Trabulo, S.; Cardoso, A.L.; Mano, M.; De Lima, M.C.P. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals 2010, 3, 961–993.
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836.
- Nakase, I.; Tanaka, G.; Futaki, S. Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells. Mol. BioSyst. 2013, 9, 855.
- Pan, R.; Xu, W.; Ding, Y.; Lu, S.; Chen, P. Uptake Mechanism and Direct Translocation of a New CPP for siRNA Delivery. Mol. Pharm. 2016, 13, 1366–1374.
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a bio-logically active protein into the mouse. Science 1999, 285, 1569–1572.
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In vivo delivery of a Bcl-xL fusion protein containing the TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J. Neurosci. 2002, 22, 5423–5431.
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964.
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185.