Iimmunotherapy has emerged as a standard-of-care for most human malignancies, including head and neck cancer, but only a limited number of patients exhibit a durable clinical benefit. An urgent medical need is the establishment of accurate response predictors, which is handicapped by the growing body of molecular, cellular and clinical variables that modify the complex nature of an effective anti-tumor immune response.
- Head and Neck Cancer
1. Introduction
Head and neck cancers (HNCs) are among the most frequent and destructive human cancers worldwide, causing considerable morbidity and mortality [1][2]. Head and neck squamous cell carcinomas (HNSCCs) account for the majority of HNC, are unexpectedly heterogeneous in nature, and tobacco use, extensive alcohol consumption and infection with high-risk human papillomavirus (HPV), in particular HPV16, are the main etiological risk factors [3][4][5]. As for many other solid cancers, HNSCC pathogenesis resembles a tightly orchestrated balance between immune effector response and tolerance, and cancer cells evade the host immune surveillance by a broad range of cellular and molecular mechanisms [6][7][8]. Hence, the tumor immune microenvironment (TIME) of individual HNSCCs is rather heterogeneous and characterized by a broad spectrum of qualitative and quantitative differences in immune cell populations [9].
Despite continuous improvement in conventional treatments, consisting of surgery, radio- and chemotherapy, a substantial proportion of HNSCC patients suffer from locoregional relapse or distant metastasis [3][10]. For patients with recurrent or metastatic disease (R/M-HNSCC) the armamentarium of systemic anti-cancer modalities and innovative local approaches continues to grow, but the overall survival remains dismal and is still unsatisfactory [11][12]. In the past decade, immunotherapy based on immune checkpoint inhibition (ICI) has become an essential pillar for cancer treatment and now represents the standard of care for most human cancers, including HNSCC [3][13]. Activation of immune checkpoint cascades such as those controlled by cytotoxic T lymphcytes associated protein 4 (CTLA-4) or programmed cell death protein 1 (PD-1) and its ligand (PD-L1) results in inactivation of tumor-specific T cells and immune evasion. The underlying concept of ICI is that treatment with anti-PD-(L)1 or anti-CTLA-4 antibodies reinvigorates cytotoxic immune cells to target cancer cells [14][15][16].In randomized phase III trials two monoclonal antibodies (nivolumab and pembrolizumab), targeting PD-1 demonstrated longer overall survival in comparison with standard chemotherapy in pretreated R/M-HNSCC [17][18].Furthermore, pembrolizumab demonstrated superiority relative to standard first-line cytotoxic treatment for R/M-HNSCC, either as single-agent therapy or in combination with chemotherapy for tumors with PD-L1 expression [19].However, a major limitation is the low overall response rate of ICI therapy and many patients have experienced minimal or no clinical benefit [13][20]. Due to the relatively poor response rate, potential risk for hyper-progressive disease, and high degree of immune-related adverse events (irAEs), an urgent medical demand exists for reliable cellular or molecular biomarkers (
Figure 1) to support treatment-decision making and a better stratification of cancer patients at higher risk for intrinsic or acquired treatment failure, who might benefit from new strategies of combination therapies [21][22].
Figure 1.
2. Immune-Related Mutational and Epigenetic Landscapes
Figure 3). Somatic mutations in tumor DNA can induce neoantigens production, which can be targeted and recognized by the immune system, particularly after treatment with agents that activate T cells [23]. Those somatic mutations are transcribed and translated, and neoantigen-containing peptides are processed by the antigen-processing machinery and are loaded onto MHC molecules to be presented on the cell surface. However, not all somatic mutations produce peptides which are appropriately processed and loaded onto MHC complexes, and even fewer can be recognized by T cells [7][24][25]. Recent studies have revealed mutational signatures underlying the evolution of cancer and highlighted a strong association of HPV with APOBEC mutational signatures, suggesting impaired antiviral defense as a driving force in distinct cancers, including HNSCC [24][25][26]. Almost all HPV-positive and many HPV-negative HNSCCs share a large fraction of somatic mutations attributable to members of the apolipoprotein B mRNA editing enzyme catalytic subunit-like protein 3 (APOBEC3) family of single-stranded DNA cytosine deaminases [27][28]. Utilizing whole-exome and RNA-seq datasets from The Cancer Genome Atlas (TCGA), Faden et al. [29] observed the highest IFN-γ levels for HNSCC across cancer types with high APOBEC-related mutational burden. Most prominent IFN-γ scores in HNSCC were present in HPV-related tumors and tumor-specific neoantigens were significantly correlated with mutational burden attributed to APOBEC [29]. In another study, a subgroup of APOBEC-enriched, HPV-negative HNSCC with a distinct immunogenic phenotype was identified, which was characterized by higher T-cell inflammation, prominent immune checkpoint expression and enrichment of mutations in immune-evasion pathways [30].
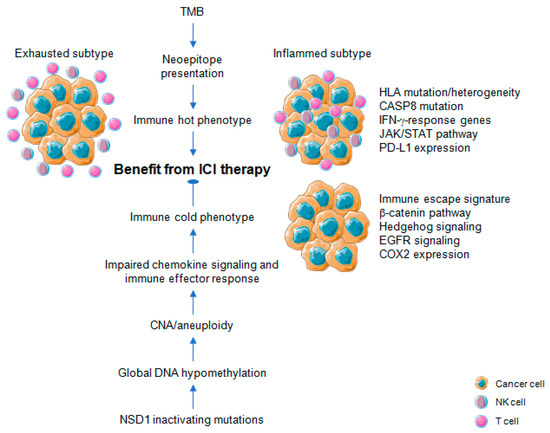
Figure 3.
Figure 3). A higher burden of copy number loss in non-responders to CTLA-4 and PD-1 blockade was identified in a cohort of melanoma patients, which was associated with decreased expression of genes in immune-related pathways [31]. Davoli et al. [32] investigated 12 cancer types from TCGA to demonstrate that most highly aneuploid tumors exhibit reduced expression of markers for infiltrating immune cells, especially CD8-positive T cells and NK cells, indicating that aneuploidy restricts cytotoxic immune response during tumorigenesis. Again, tumor aneuploidy inversely correlates with patient survival in two clinical trials of ICI therapy for metastatic melanoma [32]. The effect on treatment response for TMB and aneuploidy was non-redundant, suggesting that both alterations reflect different aspects in the balance of immune surveillance versus tolerance and the potential utility of a combinatorial biomarker to optimize patient care with ICI therapy [31]. In line with this assumption, the combination of TMB and CNA for prognostic risk assessment and prediction of heterogeneous clinical responses to ICI treatment was confirmed for multiple cancers [33][34]. A high level of aneuploidy was also found for an immune cold subtype with least amount of tumor infiltrating lymphocytes (TILs) based on a pan-SCC cohort of TCGA [35]. For primary HNSCC, an inverse correlation between copy number alteration and measures of immune infiltration was evident [36][37], and a lower cytotoxic immune phenotype exhibited a characteristic pattern of copy number loss affecting chemokine signaling and immune effector response [38].
Profound global loss of DNA methylation is a hallmark of many cancers, and global demethylation in cancer promotes chromosomal instability [39][40], particularly involving large-scale alterations leading to aneuploidy [41]. Cancers commonly hijack various epigenetic mechanisms to escape immune restriction, but the impact of DNA methylation on immune evasion and in the context of cancer immunotherapy has been addressed only recently (
Figure 3) [42][43][44]. In a pan-cancer analyses of TCGA data, Jung et al. [45] found that genomic hypomethylation correlated not only with aneuploidy but also immune escape signatures independently of the mutational burden, and was associated with increased immunotherapeutic resistance. Moreover, inactivating mutations in the nuclear receptor binding SET domain protein 1 (NSD1) a histone methyltransferase define an intrinsic subtype of HNSCC that features pronounced DNA hypomethylation [46][47][48] and displays an immune cold phenotype characterized by low levels of TILs and low expression of a CD8-positive T cell inflamed gene signature [49][50]. These data indicate NSD1 as a tumor cell-intrinsic driver of an immune cold phenotype by causing epigenetic deregulation with potential implications for immunotherapy (
Reactivation of transposable elements (TEs) including endogenous retroviral (ERV) transcripts is another consequence of profound global loss of DNA methylation in cancer. It results in a state of viral mimicry in which treated cancer cells mount an immune response by turning on viral defense genes and potentially expressing neoantigens [51]. In a pan-cancer analysis with TCGA cohorts, expression of 262 TE subfamilies appear to result from a proximal loss of DNA methylation [52]. TE overexpression in tumor samples with respect to matched normal controls is most prominent in stomach, bladder, and liver cancer as well as HNSCC. At the global level, this overexpression in HNSCC is associated with loss of DNA methylation, particularly at proximal CpG sites, suggesting targeted loss of DNA methylation near TE sites as a major mode of regulation [52]. For HNSCC, tumors with a high ERV expression pattern share prominent immune checkpoint pathway activation and increased immune infiltration with a higher CD8-positive T cell fraction as compared with ERV low expressing counterparts [53]. These data together with recent preclinical studies provide a strong rational for combining epigenetic targeting and immune checkpoint blockade in HNSCC to enhance treatment efficacy. Due to pleiotropic effects on multiple targets, which could limit the risk for treatment resistance, inhibition of epigenetic modifications emerges as promising strategy in combination with ICI [54]. Indeed, increased TE expression and de novo presentation of TE-derived peptides on MHC class I molecules were found upon treatment of cancer cells with a demethylation agent, indicating that therapeutic reactivation of tumor-specific TEs may synergize with immunotherapy. In line with this assumption, the phase IbNIBIT-M4 trial reported that treatment of patients with advanced melanoma using the next-generation DNA hypomethylating agent guadecitabine combined with ipilimumab is safe and tolerable, and shows promising immunomodulatory and antitumor activity [55].However, in an open-label phase II multi-cohort study administration of the oral DNA hypomethylating agent CC-486 combined with durvalumab did not demonstrate robust pharmacodynamic or clinical activity in selected immunologically cold solid tumors consisting of PD-(L)1 inhibitor naïve patients with either advanced microsatellite stable colorectal cancer, platinum resistant ovarian cancer, or estrogen receptor positive, HER2 negative breast cancer [56].
Enhancer of zeste homolog 2 (EZH2), a methyltransferase subunit of the polycomb repressive complex 2 (PRC2) that catalyzes histone H3 methylation on lysine 27 (H3K27), represents another epigenetic target to circumvent ICI resistance in HNSCC [57][58]. EZH2 expression was negatively correlated with components of the antigen-processing machinery pathway in TCGA-HNSC and genetic ablation or pharmacological inhibition of EZH2 resulted in a significant increase of MHC class I expression on HNSCC cells, antigen-specific CD8-positive T cell proliferation, IFN-γ production, and tumor cell cytotoxicity. In a preclinical mouse model, the combination of an EZH2 inhibitor (GSK126) and anti-PD1 antibodies suppressed tumor growth of anti-PD-1-resistant HNSCC [58]. The association of chromatin modification with CD8-positive T cell exclusion in HPV-negative HNSCC was further supported by a study of Vougiouklakis et al. [59]. They identified a couple of protein methyltransferases (PMTs) and demethylases (PDMTs) with inverse expression pattern compared to components of the antigen presentation machinery, CD8-positive T cells and immune-active chemokines. Finally, a phase II trial of pembrolizumab and vorinostat, a pan-HDAC (histone deacetylase) inhibitor, with progressing and incurable head and neck cancers demonstrated activity in R/M-HNSCC, but fewer responses in salivary gland cancer [60]. However, toxicities were higher than reported with pembrolizumab alone and no complete responder was observed.