Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated receptors/transcriptional factors composed by three distinct isoforms called PPARα (nuclear receptor subfamily 1 group C, NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3), each of which is encoded by independent genes in rodents and humans.
- peroxisome proliferator-activated receptors (PPARs)
- endocannabinoid system (ECS)
1. PPAR Receptors Classification, Distribution, and Function
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated receptors/transcriptional factors composed by three distinct isoforms called PPARα (nuclear receptor subfamily 1 group C, NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3), each of which is encoded by independent genes in rodents and humans. Although co-expressed in several different types of organs and tissues, each isoform shows distinctive functional characteristics and ligand specificity [1,2][1][2]. Since their identification, PPARs have been recognized as sensing receptors for a variety of endogenous lipids (such as unsaturated, monounsaturated, and poly-unsaturated fatty acids) and natural exogenous compounds.
1.1. PPARα
PPARα is highly expressed in organs and tissues characterized by a high rate of fatty acid catabolism for energy production including liver, brown adipose tissue, endocrine tissues, gastrointestinal tract, cardiac and skeletal muscle. To a lesser extent, PPARα is present in the kidney, adrenal tissues, endothelial and immune (i.e. macrophages, monocytes, and lymphocytes) cells [3,4][3][4]. Additionally, PPARα has been found in specific brain areas where it exerts anti-inflammatory and neuroprotective actions (see next section) [5,6,7,8,9][5][6][7][8][9].
1.1.1. Role of PPARα in Metabolism
PPARα plays a major role in metabolic homeostasis regulating lipid metabolism. Specifically, during the fed-to-fasted transition, PPARα drives the production of enzymes responsible for fatty acid oxidation (FAO) and the synthesis of ketone bodies from fatty acids in the liver. Thereby, PPARα works as a hub that integrates multiple metabolic signals to orchestrate the switch from glucose to fatty acid utilization for energy production [10,11][10][11]. Additionally, PPARα governs hepatic amino acid metabolism [12]. Studies on PPARα in other tissues including the heart, small intestine, skeletal muscle and brain have indicated that the role of PPARα in metabolic homeostasis is well conserved between different cell types [12,13,14,15][12][13][14][15]. Interestingly, Pparα KO mice, under a normal dietary regimen, do not display pronounced anomalies in their phenotype. However, under fasting conditions or a high-fat diet, Pparα KO mice develop hypoglycemia and dyslipidemia characterized by excessive production of triacylglycerols [13,16,17][13][16][17]. Moreover, the lack of PPARα causes cardiac metabolic and contractile dysfunction, morphological and functional alterations in the brain in association with micro-/macro- vasculature dysfunctions [15,18,19][15][18][19]. In general, although the Pparα KO mouse model still shows unresolved aspects that are most likely attributable to the compensatory role of the other two PPAR isotypes, it allowed a deeper understanding of the role of PPARα in energy metabolism.
1.1.2. Neuroprotective and Anti-Inflammatory Role of PPARα
Although all three PPAR isoforms are expressed in the nervous system during embryogenesis, only PPARβ/δ expression remains high in the brain, whereas PPARα and PPARγ expression decreases postnatally and remain restricted to the specific brain areas [20]. In particular, PPARα is expressed in basal ganglia, the reticular formation, some thalamic, mesencephalic and cranial motor nuclei and the large motoneurons of the spinal cord [5]. PPARα is expressed in dopamine neurons of the substantia nigra and spiny neurons of the dorsal striatum where it decreases dopaminergic transmission thus exerting an important control in behavioural responses [8,21,22][8][21][22]. The expression of PPARα is also reported in different subfields of the hippocampus of rodents, where it is also involved in the control of neuronal excitability and synaptic plasticity via cyclic AMP response element-binding protein (CREB) [23]. Moreover, PPARα is present in oligodendrocytes, microglia and astrocytes [24,25,26][24][25][26]. The studies reported above as well many others suggest that activation of PPARα in the brain initiates anti-oxidative and anti-inflammatory processes that confer neuroprotection. An effect also exerted by preserving the microvasculature activity [27]. The neuroprotective role of PPARα has been documented in numerous studies using several genetic and/or pharmacological models of neurodegenerative disorders including stroke, Alzheimer’s disease, Parkinson’s disease, traumatic brain injury, diabetic peripheral neuropathy, and retinopathy [7,9,22,28,29][7][9][22][28][29]. Besides its neuroprotective role, PPARα exerts important anti-inflammatory effects also in peripheral organs and tissues [3,30][3][30]. In this regard, Luisa et al., 2009, using an experimental model of inflammatory bowel disease, showed that Pparα KO mice compared to wild-type (WT) ones, have a deficient anti-inflammatory response [31]. Additionally, PPARα agonists have been reported to exert anti-inflammatory, antipyretic, anti-atherogenic and analgesic effects as demonstrated in experimental models of spinal cord trauma [28[28][32],32], neuropathic pain [33,34,35][33][34][35] and high-fat diet (HFD) induced atherosclerosis [36].
1.2. PPARγ
PPARγ is considered a master regulator of adipogenesis and is abundantly expressed in adipose tissue where it is primarily involved in fat and carbohydrate metabolism. Additionally, PPARγ exerts anti-inflammatory effects in several types of tissues and organs by repressing the expression and function of pro-inflammatory factors such as NF-κB and Nrf2/CREB and it is implicated in cancer and atherosclerosis [32]. The expression of PPARγ was also reported in other organs and tissues including the liver, skeletal muscle, spleen, heart, placenta, lung, ovary, and also brain (glial cells and neurons) [33,34][33][34].
1.2.1. Role of PPARγ in Metabolism
In both rodents and humans, PPARγ is a master regulator of adipocytes differentiation as well as glucose and lipid metabolism [37]. Additional actions of PPARγ include the regulation of adipokines and inflammatory mediators expression, M1/M2 macrophage polarization, atherosclerosis and bone formation [38,39][38][39]. Genetic ablation of PPARγ in mice is lethal [40]. However, in an elegant study conducted by Gavrilova et al. [41], it was demonstrated that young mice with a PPARγ-deficient liver show a similar profile of wild-type (wt) mice. However, with ageing, LPPARγ Knock-Out (Lpparγ-KO) mice develop fat intolerance, increased adiposity, hyperlipidemia, and insulin resistance. Interestingly, when fed with a lipogenic diet even young LPPARγ-KO mice developed obesity. Other studies showed that mutant PPARγ mice show a complex metabolic phenotype including increased lean mass with organomegaly, hypermetabolism, hyperphagia, and lipoatrophy [42,43,44][42][43][44]. Moreover, Lüdtke et al. reported that PPARγ mutations lead to a familial form of lipodystrophy [45]. Therefore, these findings provide evidence that PPARγ is a key target to protect the liver as well as other organs and tissues from fat accumulation, insulin resistance, and uncontrolled inflammatory responses. Interestingly, the PPARγ gene is characterized by distinct mRNA isoforms due to alternative splicing of five exons at the 5′-terminal regions (A1, A2, B, C, and D). In particular, among the seven distinct isoforms identified so far, the most known are PPAR-γ1, -γ2, and –γ3. The PPARγ1 mRNA isoform is expressed in a wide range of organs and tissues including those aforementioned and also in the immune cells (e.g., monocytes/macrophages). On the contrary, the expression of PPARγ2 mRNA seems restricted to adipose tissue, and PPARγ3 is localized in macrophages, colon, and adipose tissue [37,46,47,48,49][37][46][47][48][49]. Of note, in 2004 Zhang et al. showed that PPARγ2 deficient mice (PPARγ -/-) have an impaired fat metabolism and insulin resistance, thus demonstrating the central role of PPARγ2 in adipogenesis [14]. It is important to recall that different PPARγ isoforms may be responsible for unique tissue-specific biological effects in response or not to endogenous and/or exogenous ligands.
1.2.2. Neuroprotective and Anti-Inflammatory Role of PPARγ
PPARγ, similarly to PPARα, in the brain exerts anti-inflammatory and neuroprotective effects through the inhibition of NFkB, AP-1, STATs, and iNOS [50]. This finding has been confirmed in several animal models of Alzheimer’s and Parkinson’s disease. Moreover, the synthetic PPARγ agonist pioglitazone was shown to extend the survival of SOD1-G93A, a mouse model of Amyotrophic lateral sclerosis (ALS), by delaying the onset of the disease and preventing the death of motor neuron cells [51]. Activation of PPARγ has been shown to exert neuroprotective effects also by preventing astro- and microglial activation [52]. Additionally, PPARγ has been shown to downregulate the expression of pro-inflammatory factors and free radical production in various intestinal disorders including colon cancer, gastritis and irritable bowel syndrome, and to also exert anti-nociceptive effects against somatic pain, allergic and skin diseases [53,54,55][53][54][55].
1.3. PPARβ/δ
PPARβ/δ is ubiquitously expressed and its biological functions mainly overlap with those of the other two PPAR isoforms. Recently, the expression of PPARβ/δ was demonstrated in neurons, astrocytes, oligodendrocytes, and also, in microglial cells [56,57][56][57]. Several studies demonstrate that PPARβ/δ plays an important role in inflammatory processes and, due to its proangiogenic and anti-/pro-carcinogenic properties, it is considered a therapeutic target for treating metabolic syndrome, dyslipidemia, diabetes while the role of PPARβ/δ on cancerogenesis is still debated [58,59][58][59]. In the brain, PPARβ/δ could act as a protective factor for counteracting inflammation and promoting antioxidant mechanisms [60]. Additionally, PPARβ/δ is known to control cell cycle, proliferation, differentiation and also inhibit cell death by promoting the expression of ILK and PDK1 in many types of cells [61,62][61][62]. However, compared to the other two isoforms, the role of PPARβ/δ in inflammation still needs to be fully elucidated. For example, at peripheral level, stimulation of PPARβ/δ was shown to promote the inhibition of inflammatory response in streptozotocin-induced diabetic nephropathy and vasculopathy [63,64][63][64]. In contrast, other studies report that PPARβ/δ appears to promote inflammation in other contexts. This latter evidence was observed in murine models of skin disorders (i.e. psoriasis) and arthritis [65].
2. Structure of PPARs
Like other nuclear receptor superfamily members, PPAR structure is organized in four domains, named A/B, C, D, and E/F: (i) the N-terminal A/B domain contains the ligand-independent activation function 1 (AF1) responsible for the transcriptional activation; (ii) the C domain consists in the DNA-binding domain (DBD), formed by two zinc-finger motifs responsible for the binding to peroxisome proliferator response elements (PPREs) within the promoter regions of target genes; (iii) the D domain (hinge domain) modulates the ability of the receptor to bind DNA and it is involved in corepressors binding; (iv) C-terminal E/F domains correspond to the ligand-binding domain (LBD) which contains the ligand-binding pocket (LBP), the ligand-dependent activation function (AF-2) and regions important for the heterodimerization with the retinoid X receptor (RXR) [66,67][66][67] (Figure 1A). The PPAR-RXR heterodimer is a prerequisite for PPARs to bind the PPREs in the promoter region of target genes. They act as permissive heterodimers, being activated either by PPAR or RXR ligands. However, the simultaneous presence of both ligands gives rise to a synergistic response [68]. When PPAR-RXR heterodimers are not bound to a ligand, they act as repressors through association with corepressor complexes (Figure 1B,C; see also Figure 2). The ligand binding to LBP induces conformational changes in the AF2 region, facilitating the recruitment of coactivators and the release of corepressors [69]. In detail, canonical agonists within the LBP form a network of hydrogen bonds stabilizing the conformation of a short helix, the helix12 (H12), which in turn allows the interaction of coactivators with the AF-2 surface. However, PPAR ligands can also bind to distinct sub-regions or allosteric sites and activate these receptors through H12-independent mechanisms to elicit a partial agonist activity [70]. Besides the ligand-dependent mechanism, other mechanisms are known to regulate the transactivation and trans-repression activity of PPARs. In particular, the PPAR activity is governed by various post-translational modifications including phosphorylation, SUMOylation, ubiquitination, acetylation, and O-GlcNAcylation [71].
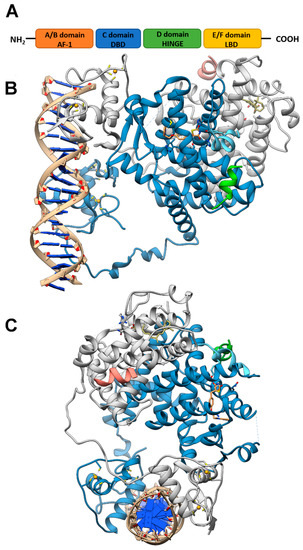
Figure 1. PPAR domains and 3D model of their interaction with RXRα and target DNA (A) Schematic representation of the PPAR domain organization and (B,C) orthogonal views of the x-ray structure of PPARγ/RXRα heterodimer in complex with DNA and coactivator peptides (PDB id: 3DZY). PPARγ is colored in slate blue with helix H12 in sky blue, RXRα in gray, the coactivator peptides in green and salmon and DNA in tan. The ligands rosiglitazone and 9-cis-retinoic acid are shown in rose brown and khaki, respectively, while the Zn(II) ions are in gold. Nitrogen, oxygen and sulfur atoms are colored in blue, red and yellow, respectively. This figure has been realized with the Chimera v.1.14 program.
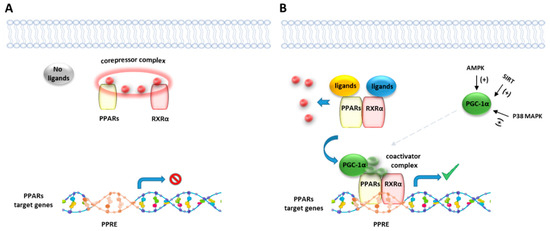
Figure 2. Representative illustration of PPARs activating pathways. (A) In absence of ligand binding, the nuclear corepressor complex prevents PPAR/RXR binding to their DNA binding element, called PPAR response element (PPRE). (B) In presence of a ligand, PPAR/RXR dimer binds to the PPRE of target genes and regulates the transcription of target genes. Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is a transcriptional coactivator of the PPAR/RXR complex, known to be activated by intracellular factors such as AMPK, SIRT and MAP kinases.
References
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential Expression and Activation of a Family of Murine Peroxisome Proliferator-Activated Receptors. PNAS 1994, 91, 7355–7359.
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome Proliferator-Activated Receptor Structures: Ligand Specificity, Molecular Switch and Interactions with Regulators. Biochim. Biophys. Acta 2007, 1771, 915–925.
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in Health and Disease. Nature 2000, 405, 421–424.
- Takeyama, K.; Kodera, Y.; Suzawa, M.; Kato, S. [Peroxisome proliferator-activated receptor(PPAR)--structure, function, tissue distribution, gene expression]. Nihon Rinsho 2000, 58, 357–363.
- Moreno, S.; Farioli-Vecchioli, S.; Cerù, M.P. Immunolocalization of Peroxisome Proliferator-Activated Receptors and Retinoid X Receptors in the Adult Rat CNS. Neuroscience 2004, 123, 131–145.
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of Anti-Inflammatory and Neuroprotective Actions of PPAR-Gamma Agonists. Front. Biosci. 2008, 13, 1813–1826.
- Melis, M.; Carta, S.; Fattore, L.; Tolu, S.; Yasar, S.; Goldberg, S.R.; Fratta, W.; Maskos, U.; Pistis, M. Peroxisome Proliferator-Activated Receptors-Alpha Modulate Dopamine Cell Activity through Nicotinic Receptors. Biol. Psychiatry 2010, 68, 256–264.
- Melis, M.; Scheggi, S.; Carta, G.; Madeddu, C.; Lecca, S.; Luchicchi, A.; Cadeddu, F.; Frau, R.; Fattore, L.; Fadda, P.; et al. PPARα Regulates Cholinergic-Driven Activity of Midbrain Dopamine Neurons via a Novel Mechanism Involving Α7 Nicotinic Acetylcholine Receptors. J. Neurosci. 2013, 33, 6203–6211.
- Wójtowicz, S.; Strosznajder, A.K.; Jeżyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988.
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; März, W.; Tafuri, S.; Wahli, W.; et al. PPARα Governs Glycerol Metabolism. J. Clin. Investig. 2004, 114, 94–103.
- Peeters, A.; Baes, M. Role of PPARα in Hepatic Carbohydrate Metabolism. PPAR Res. 2010, 2010.
- Kersten, S. Integrated Physiology and Systems Biology of PPARα. Mol. Metab. 2014, 3, 354–371.
- Peters, J.M.; Hennuyer, N.; Staels, B.; Fruchart, J.C.; Fievet, C.; Gonzalez, F.J.; Auwerx, J. Alterations in Lipoprotein Metabolism in Peroxisome Proliferator-Activated Receptor Alpha-Deficient Mice. J. Biol. Chem. 1997, 272, 27307–27312.
- Zhang, J.; Fu, M.; Cui, T.; Xiong, C.; Xu, K.; Zhong, W.; Xiao, Y.; Floyd, D.; Liang, J.; Li, E.; et al. Selective Disruption of PPARgamma 2 Impairs the Development of Adipose Tissue and Insulin Sensitivity. Proc. Natl. Acad. Sci. USA 2004, 101, 10703–10708.
- Guellich, A.; Damy, T.; Lecarpentier, Y.; Conti, M.; Claes, V.; Samuel, J.-L.; Quillard, J.; Hébert, J.-L.; Pineau, T.; Coirault, C. Role of Oxidative Stress in Cardiac Dysfunction of PPARalpha-/- Mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H93–H102.
- Lee, S.S.; Pineau, T.; Drago, J.; Lee, E.J.; Owens, J.W.; Kroetz, D.L.; Fernandez-Salguero, P.M.; Westphal, H.; Gonzalez, F.J. Targeted Disruption of the Alpha Isoform of the Peroxisome Proliferator-Activated Receptor Gene in Mice Results in Abolishment of the Pleiotropic Effects of Peroxisome Proliferators. Mol. Cell Biol. 1995, 15, 3012–3022.
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A Critical Role for the Peroxisome Proliferator-Activated Receptor α (PPARα) in the Cellular Fasting Response: The PPARα-Null Mouse as a Model of Fatty Acid Oxidation Disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478.
- Hu, Y.; Chen, Y.; Ding, L.; He, X.; Takahashi, Y.; Gao, Y.; Shen, W.; Cheng, R.; Chen, Q.; Qi, X.; et al. Pathogenic Role of Diabetes-Induced PPAR-α down-Regulation in Microvascular Dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 15401–15406.
- Pérez-Martín, M.; Rivera, P.; Blanco, E.; Lorefice, C.; Decara, J.; Pavón, F.J.; Serrano, A.; Rodríguez de Fonseca, F.; Suárez, J. Environmental Enrichment, Age, and PPARα Interact to Regulate Proliferation in Neurogenic Niches. Front. Neurosci. 2016, 10, 89.
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential Expression of Peroxisome Proliferator-Activated Receptors (PPARs): Tissue Distribution of PPAR-Alpha, -Beta, and -Gamma in the Adult Rat. Endocrinology 1996, 137, 354–366.
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective Activities of Palmitoylethanolamide in an Animal Model of Parkinson’s Disease. PLoS ONE 2012, 7.
- D’Agostino, G.; Cristiano, C.; Lyons, D.J.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome Proliferator-Activated Receptor Alpha Plays a Crucial Role in Behavioral Repetition and Cognitive Flexibility in Mice. Mol. Metab. 2015, 4, 528–536.
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of Cyclic AMP Response Element Binding and Hippocampal Plasticity-Related Genes by Peroxisome Proliferator-Activated Receptor α. Cell Rep. 2013, 4, 724–737.
- Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Agonists for the Peroxisome Proliferator-Activated Receptor-Alpha and the Retinoid X Receptor Inhibit Inflammatory Responses of Microglia. J. Neurosci. Res. 2005, 81, 403–411.
- Bernardo, A.; Bianchi, D.; Magnaghi, V.; Minghetti, L. Peroxisome Proliferator-Activated Receptor-Gamma Agonists Promote Differentiation and Antioxidant Defenses of Oligodendrocyte Progenitor Cells. J. Neuropathol. Exp. Neurol. 2009, 68, 797–808.
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.E.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R.; et al. Palmitoylethanolamide Induces Microglia Changes Associated with Increased Migration and Phagocytic Activity: Involvement of the CB2 Receptor. Sci. Rep. 2017, 7, 375.
- Moran, E.P.; Ma, J. Therapeutic Effects of PPARα on Neuronal Death and Microvascular Impairment. Available online: (accessed on 24 January 2021).
- Esposito, E.; Rinaldi, B.; Mazzon, E.; Donniacuo, M.; Impellizzeri, D.; Paterniti, I.; Capuano, A.; Bramanti, P.; Cuzzocrea, S. Anti-Inflammatory Effect of Simvastatin in an Experimental Model of Spinal Cord Trauma: Involvement of PPAR-α. J. Neuroinflammation 2012, 9, 81.
- D’Orio, B.; Fracassi, A.; Ceru, M.P.; Moreno, S. Targeting PPARalpha in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 345–354.
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR Receptor in Different Diseases and Their Ligands: Physiological Importance and Clinical Implications. Eur. J. Med. Chem. 2019, 166, 502–513.
- Riccardi, L.; Mazzon, E.; Bruscoli, S.; Esposito, E.; Crisafulli, C.; Di Paola, R.; Caminiti, R.; Riccardi, C.; Cuzzocrea, S. Peroxisome Proliferator-Activated Receptor-Alpha Modulates the Anti-Inflammatory Effect of Glucocorticoids in a Model of Inflammatory Bowel Disease in Mice. Shock 2009, 31, 308–316.
- Genovese, T.; Esposito, E.; Mazzon, E.; Crisafulli, C.; Paterniti, I.; Di Paola, R.; Galuppo, M.; Bramanti, P.; Cuzzocrea, S. PPAR-Alpha Modulate the Anti-Inflammatory Effect of Glucocorticoids in the Secondary Damage in Experimental Spinal Cord Trauma. Pharmacol. Res. 2009, 59, 338–350.
- Wen, W.; Wang, J.; Zhang, B.; Wang, J. PPARα Agonist WY-14643 Relieves Neuropathic Pain through SIRT1-Mediated Deacetylation of NF-ΚB. PPAR Res. 2020, 2020, 6661642.
- Di Cesare Mannelli, L.; D’Agostino, G.; Pacini, A.; Russo, R.; Zanardelli, M.; Ghelardini, C.; Calignano, A. Palmitoylethanolamide Is a Disease-Modifying Agent in Peripheral Neuropathy: Pain Relief and Neuroprotection Share a PPAR-Alpha-Mediated Mechanism. Med. Inflamm. 2013, 2013, 328797.
- LoVerme, J.; Russo, R.; La Rana, G.; Fu, J.; Farthing, J.; Mattace-Raso, G.; Meli, R.; Hohmann, A.; Calignano, A.; Piomelli, D. Rapid Broad-Spectrum Analgesia through Activation of Peroxisome Proliferator-Activated Receptor-Alpha. J. Pharmacol. Exp. Ther. 2006, 319, 1051–1061.
- Devi, S.; Rangra, N.K.; Rawat, R.; Alrobaian, M.M.; Alam, A.; Singh, R.; Singh, A. Anti-Atherogenic Effect of Nepitrin-7-O-Glucoside: A Flavonoid Isolated from Nepeta Hindostana via Acting on PPAR—α Receptor. Steroids 2021, 165, 108770.
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The Diverse Biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312.
- Nguyen, M.T.A.; Chen, A.; Lu, W.J.; Fan, W.; Li, P.-P.; Oh, D.Y.; Patsouris, D. Regulation of Chemokine and Chemokine Receptor Expression by PPARγ in Adipocytes and Macrophages. PLoS ONE 2012, 7, e34976.
- Yang, C.-C.; Wu, C.-H.; Lin, T.-C.; Cheng, Y.-N.; Chang, C.-S.; Lee, K.-T.; Tsai, P.-J.; Tsai, Y.-S. Inhibitory Effect of PPARγ on NLRP3 Inflammasome Activation. Theranostics 2021, 11, 2424–2441.
- Gray, S.L.; Dalla Nora, E.; Vidal-Puig, A.J. Mouse Models of PPAR-Gamma Deficiency: Dissecting PPAR-Gamma’s Role in Metabolic Homoeostasis. Biochem. Soc. Trans. 2005, 33, 1053–1058.
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver Peroxisome Proliferator-Activated Receptor γ Contributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J. Biol. Chem. 2003, 278, 34268–34276.
- Kintscher, U.; Law, R.E. PPARgamma-Mediated Insulin Sensitization: The Importance of Fat versus Muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E287–E291.
- Amin, R.H.; Mathews, S.T.; Camp, H.S.; Ding, L.; Leff, T. Selective Activation of PPARgamma in Skeletal Muscle Induces Endogenous Production of Adiponectin and Protects Mice from Diet-Induced Insulin Resistance. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E28–E37.
- Gilardi, F.; Winkler, C.; Quignodon, L.; Diserens, J.-G.; Toffoli, B.; Schiffrin, M.; Sardella, C.; Preitner, F.; Desvergne, B. Systemic PPARγ Deletion in Mice Provokes Lipoatrophy, Organomegaly, Severe Type 2 Diabetes and Metabolic Inflexibility. Metabolism 2019, 95, 8–20.
- Lüdtke, A.; Buettner, J.; Schmidt, H.H.; Worman, H.J. New PPARG Mutation Leads to Lipodystrophy and Loss of Protein Function That Is Partially Restored by a Synthetic Ligand. J. Med. Genet. 2007, 44, e88.
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.-M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.-C.; Deeb, S.; et al. The Organization, Promoter Analysis, and Expression of the Human PPARγ Gene. J. Biol. Chem. 1997, 272, 18779–18789.
- Zhou, J.; Wilson, K.M.; Medh, J.D. Genetic Analysis of Four Novel Peroxisome Proliferator Activated Receptor-Gamma Splice Variants in Monkey Macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 274–283.
- Subbarayan, V.; Sabichi, A.L.; Kim, J.; Llansa, N.; Logothetis, C.J.; Lippman, S.M.; Menter, D.G. Differential Peroxisome Proliferator-Activated Receptor-Gamma Isoform Expression and Agonist Effects in Normal and Malignant Prostate Cells. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1710–1716.
- Chen, Y.; Jimenez, A.R.; Medh, J.D. Identification and Regulation of Novel PPAR-Gamma Splice Variants in Human THP-1 Macrophages. Biochim. Biophys. Acta 2006, 1759, 32–43.
- Daynes, R.A.; Jones, D.C. Emerging Roles of PPARs in Inflammation and Immunity. Nat. Rev. Immunol. 2002, 2, 748–759.
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome Proliferator-Activated Receptor-Gamma Agonist Extends Survival in Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2005, 191, 331–336.
- Heneka, M.T.; Landreth, G.E. PPARs in the Brain. Biochim. Biophys. Acta (BBA) Mol.Cell Biol. Lipids 2007, 1771, 1031–1045.
- Roudsari, N.M.; Lashgari, N.-A.; Zandi, N.; Pazoki, B.; Momtaz, S.; Sahebkar, A.; Abdolghaffari, A.H. PPARγ: A Turning Point for Irritable Bowel Syndrome Treatment. Life Sci. 2020, 257, 118103.
- Tachibana, M.; Wada, K.; Katayama, K.; Kamisaki, Y.; Maeyama, K.; Kadowaki, T.; Blumberg, R.S.; Nakajima, A. Activation of Peroxisome Proliferator-Activated Receptor Gamma Suppresses Mast Cell Maturation Involved in Allergic Diseases. Allergy 2008, 63, 1136–1147.
- Ramot, Y.; Mastrofrancesco, A.; Camera, E.; Desreumaux, P.; Paus, R.; Picardo, M. The Role of PPARγ-Mediated Signalling in Skin Biology and Pathology: New Targets and Opportunities for Clinical Dermatology. Exp. Dermatol. 2015, 24, 245–251.
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR Isotypes in the Adult Mouse and Human Brain. Sci. Rep. 2016, 6.
- Strosznajder, A.K.; Wójtowicz, S.; Jeżyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-β/δ in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2020.
- Luquet, S.; Gaudel, C.; Holst, D.; Lopez-Soriano, J.; Jehl-Pietri, C.; Fredenrich, A.; Grimaldi, P.A. Roles of PPAR Delta in Lipid Absorption and Metabolism: A New Target for the Treatment of Type 2 Diabetes. Biochim. Biophys. Acta (BBA) Mol.Cell Biol. Lipids 2005, 1740, 313–317.
- Wagner, N.; Wagner, K.-D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133.
- Hall, M.G.; Quignodon, L.; Desvergne, B. Peroxisome Proliferator-Activated Receptor Beta/Delta in the Brain: Facts and Hypothesis. PPAR Res. 2008, 2008, 780452.
- Müller, R.; Rieck, M.; Müller-Brüsselbach, S. Regulation of Cell Proliferation and Differentiation by PPARβ/δ. PPAR Res. 2008, 2008.
- Barz, T.; Spengler, D. Peroxisome Proliferator-Activated Receptor. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin, Heidelberg, 2016; pp. 1–5. ISBN 978-3-642-27841-9.
- Matsushita, Y.; Ogawa, D.; Wada, J.; Yamamoto, N.; Shikata, K.; Sato, C.; Tachibana, H.; Toyota, N.; Makino, H. Activation of Peroxisome Proliferator-Activated Receptor Delta Inhibits Streptozotocin-Induced Diabetic Nephropathy through Anti-Inflammatory Mechanisms in Mice. Diabetes 2011, 60, 960–968.
- Cheang, W.S.; Wong, W.T.; Zhao, L.; Xu, J.; Wang, L.; Lau, C.W.; Chen, Z.Y.; Ma, R.C.W.; Xu, A.; Wang, N.; et al. PPARδ Is Required for Exercise to Attenuate Endoplasmic Reticulum Stress and Endothelial Dysfunction in Diabetic Mice. Diabetes 2017, 66, 519–528.
- Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339.
- O’Sullivan, S.E. Cannabinoids Go Nuclear: Evidence for Activation of Peroxisome Proliferator-Activated Receptors. Br. J. Pharmacol. 2007, 152, 576–582.
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res. 2015, 2015, 816856.
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266.
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-Regulated Gene Expression. PPAR Res. 2010, 2010.
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARgamma Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271.
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738.