Neural stem cells (NSCs) offer great potential for regenerative medicine due to their excellent ability to differentiate into various specialized cell types of the brain. In the central nervous system (CNS), NSC renewal and differentiation are under strict control by the regulation of the pivotal SLIT-ROBO Rho GTPase activating protein 2 (SRGAP2)—Family with sequence similarity 72 (FAM72) master gene (i.e., |-SRGAP2–FAM72-|) via a divergent gene transcription activation mechanism. If the gene transcription control unit (i.e., the intergenic region of the two sub-gene units, SRGAP2 and FAM72) gets out of control, NSCs may transform into cancer stem cells (CSCs) and generate brain tumor cells responsible for brain cancer such as glioblastoma multiforme (GBM).
- brain cancer
- cell cycle
- differentiation
- glioblastoma
- proliferation
- stem cell
1. Introduction
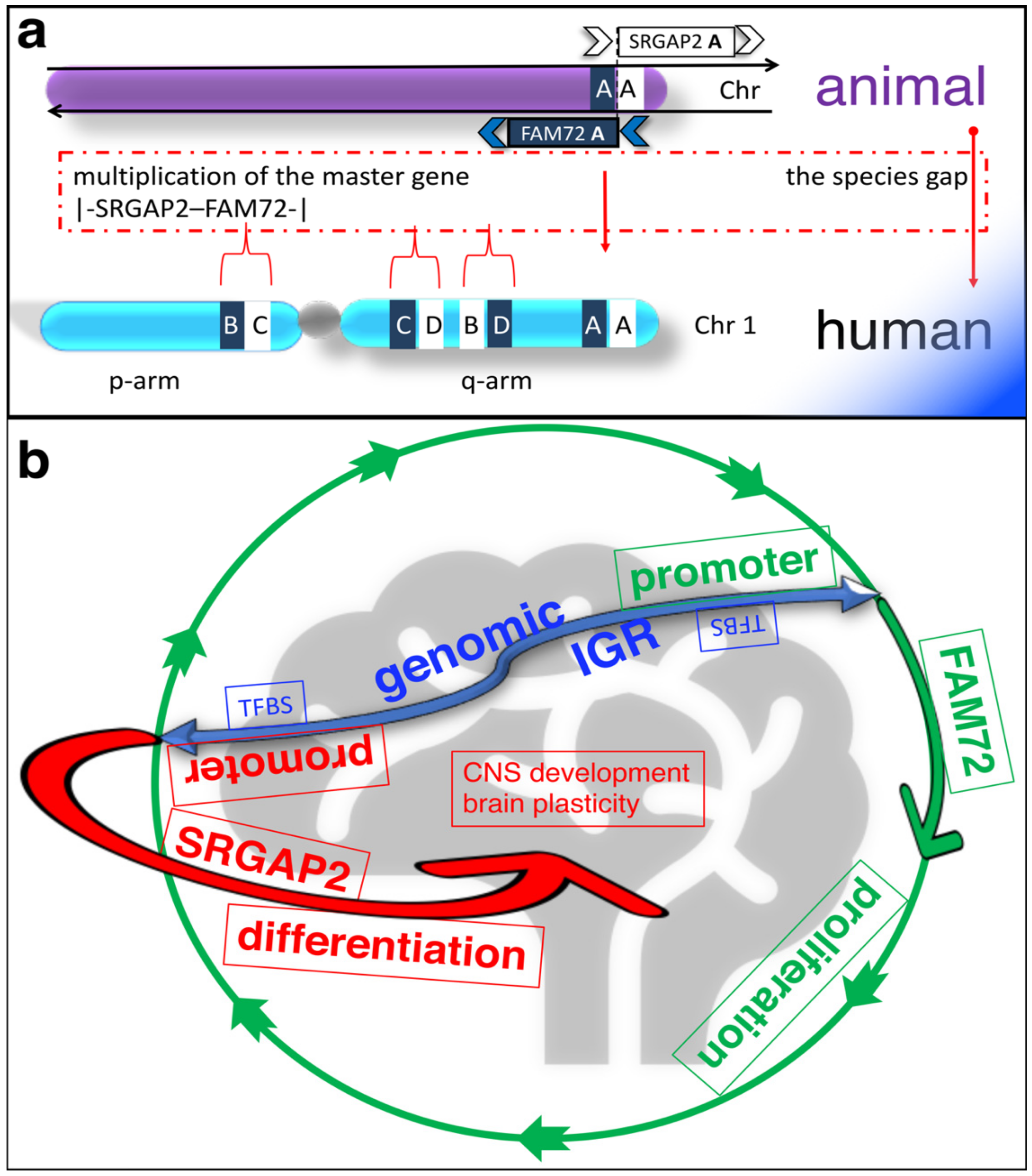
The human brain is a unique organ that can perform higher cognitive functions and is therefore different from all other species. Its uniqueness is reflected in the expression of four paralog gene pairs |-SRGAP2–FAM72-| (A–D) [1][2]. FAM72 is active in proliferating neural stem cells (NSCs) found in the brain hippocampus [1][2][3][4][5]. There are four specific FAM72 (A–D) paralogs associated with four respective SRGAP2 paralogs on human chromosome 1 (chr 1), but only one such gene pair co-exists as the |-SRGAP2–FAM72-| master gene in all other notochord containing vertebrates (Figure 1a) [1][2][6][7].
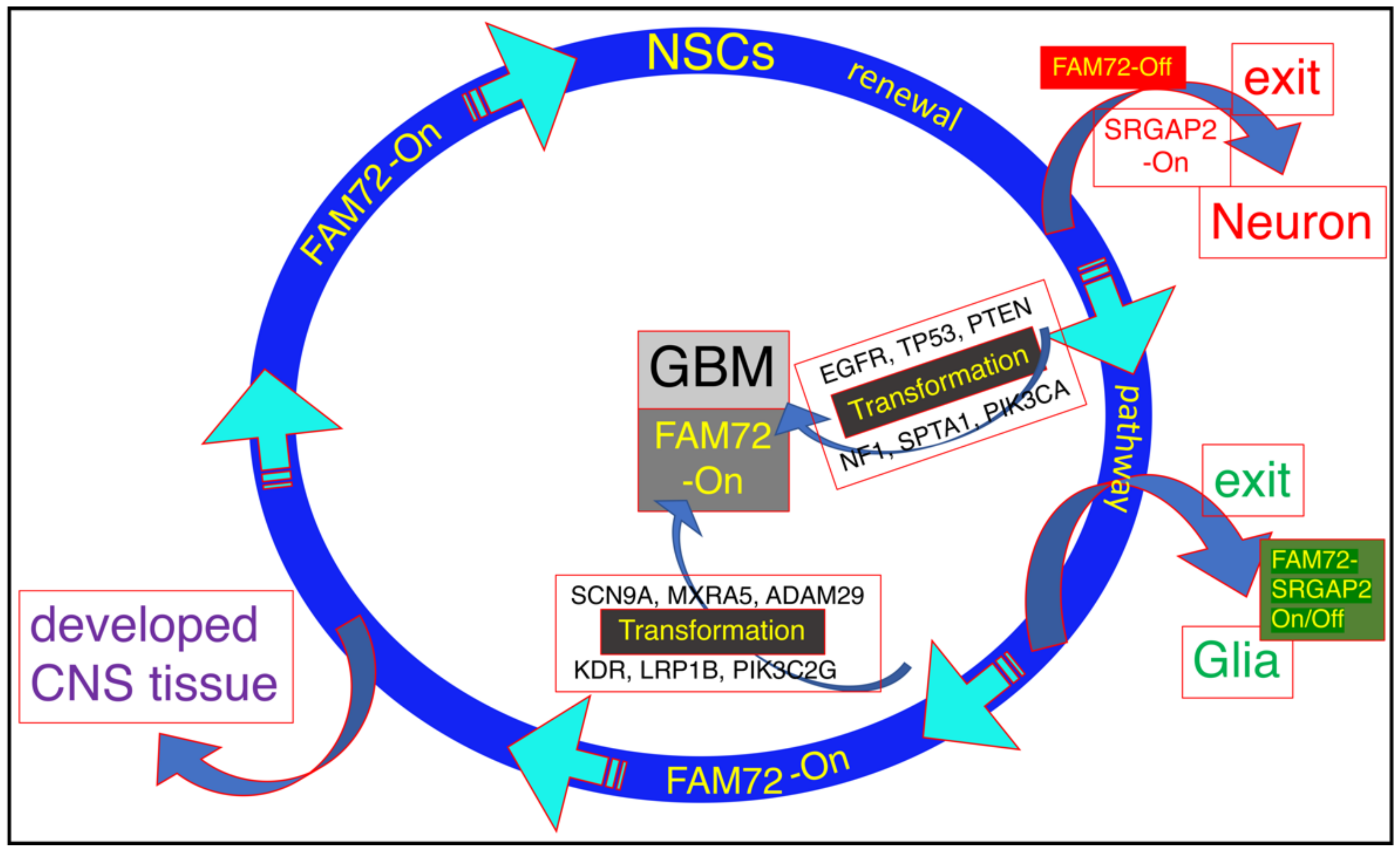
2. FAM72 and Its Role in Cancer Therapy: Therapeutic Options against Tumorigenic FAM72
Targeting FAM72 could thus be a viable treatment method for several cancer types outside the CNS because knockout of neural-specific FAM72 gene function in non-neuronal tissue may cause spindle assembly defects outside the CNS, followed by cell differentiation, senescence, or death by mitotic catastrophe in all non-neuronal proliferating cancer cells. FAM72 is an attractive target for therapy as it is a proliferative marker expressed during the late G2/M-phase of the cell cycle as well as its low expression in normal non-neuronal tissues [3][8][9], and multiple potential approaches are possible.
2.1. Therapeutic Options against Tumorigenic FAM72: RNA Interference (RNAi)
RNAi has emerged as a very effective tool for in vivo selective silencing of gene transcription, and substantial progress has been made in analyzing the therapeutic potential of various RNAi products. There are certain advantages of using RNAi for cancer therapy including the ability to target any gene including FAM72A [4], low dosages, and extended inhibition after a single dose [10]. Recently conducted clinical trials against solid tumors are promising, with the RNAi being delivered via nanoparticles [10][11]. Short hairpin-loop RNAs (shRNAs) have been demonstrated to knockdown FAM72A activity, leading to differentiation in NSCs [4]. This proves the efficacy of the approach in developing therapy against FAM72. Another approach would be to target both small interfering RNAs (siRNAs) as well as telomerase reverse transcriptase and/or MKI67 [12]. Briefly, the authors constructed adenovirus containing siRNAs targeting both MKI67 as well as the telomerase reverse transcriptase. Gene silencing for multiple oncogenes using more than one siRNA have been demonstrated before [13], and the experiment by Fang et al. [12] inhibited renal cancer cells in vitro. An oncolytic vector containing siRNAs targeted toward FAM72A as well as telomerase reverse transcriptases could prove effective without affecting normal cells, especially in non-neuronal tissues.
Another approach would be the application of antisense oligodeoxynucleotides (ASOs). ASOs are synthetically generated nucleotide sequences, about 12–25 bases long, which can be tailored according to the target sequence of interest. Intracellular binding of the ASO to its target mRNA results in RNAse cleavage, thereby leading to a lack of mRNA translation and protein formation. Currently, there are approximately 90 ongoing clinical cancer trials evaluating treatment with ASOs, with a majority being in phase I [14][15]. Animal models have proved the efficacy in inhibiting tumor formation using MKI67 ASOs, however, issues remain with optimizing dosage and nuclease degradation susceptibility [16][17]. There have been some successes using ASO cancer trials. OT-101, a phosphorothioate ASO designed for the targeted inhibition of human transforming growth factor beta 2 (TGFβ2) mRNA, has proceeded to the phase I/II clinical trial and demonstrated encouraging results [18]. AZD9150, a STAT3-inhibiting ASO, has demonstrated tumor suppressive activity in lung and lymphoma models as well as in a phase1b trial of pretreated lymphoma patients [19][20]. Another group reported that AZD9150 increases drug sensitivity and decreases tumorigenicity in neuroblastomas [21]. Recruitment for AZD9150 trials in colorectal, pancreatic, and lung cancer is ongoing (NCT02983578) [22].
Although RNAi-based drug therapeutic trials have been ongoing for some time, it was only in 2018 that the Food and Drug Administration (FDA) approved the first RNAi-based drug ONPATTRO, which is used to treat transthyretin amyloidosis. Due to a better understanding of the clinical development process required for RNAi therapeutics, more candidates are presently in development and trials, especially for cancer [23]. Selection and design of a delivery vector for RNA duplexes targeted toward FAM72 would be critical. Benayoun et al. have already demonstrated RNA silencing for FAM72, utilizing shRNA lentiviral constructs [4]. Alternatively, gRNA delivery via any of the methods above-mentioned could be performed to knockout FAM72.
2.2. Therapeutic Options against Tumorigenic FAM72: CRISPR-Cas9
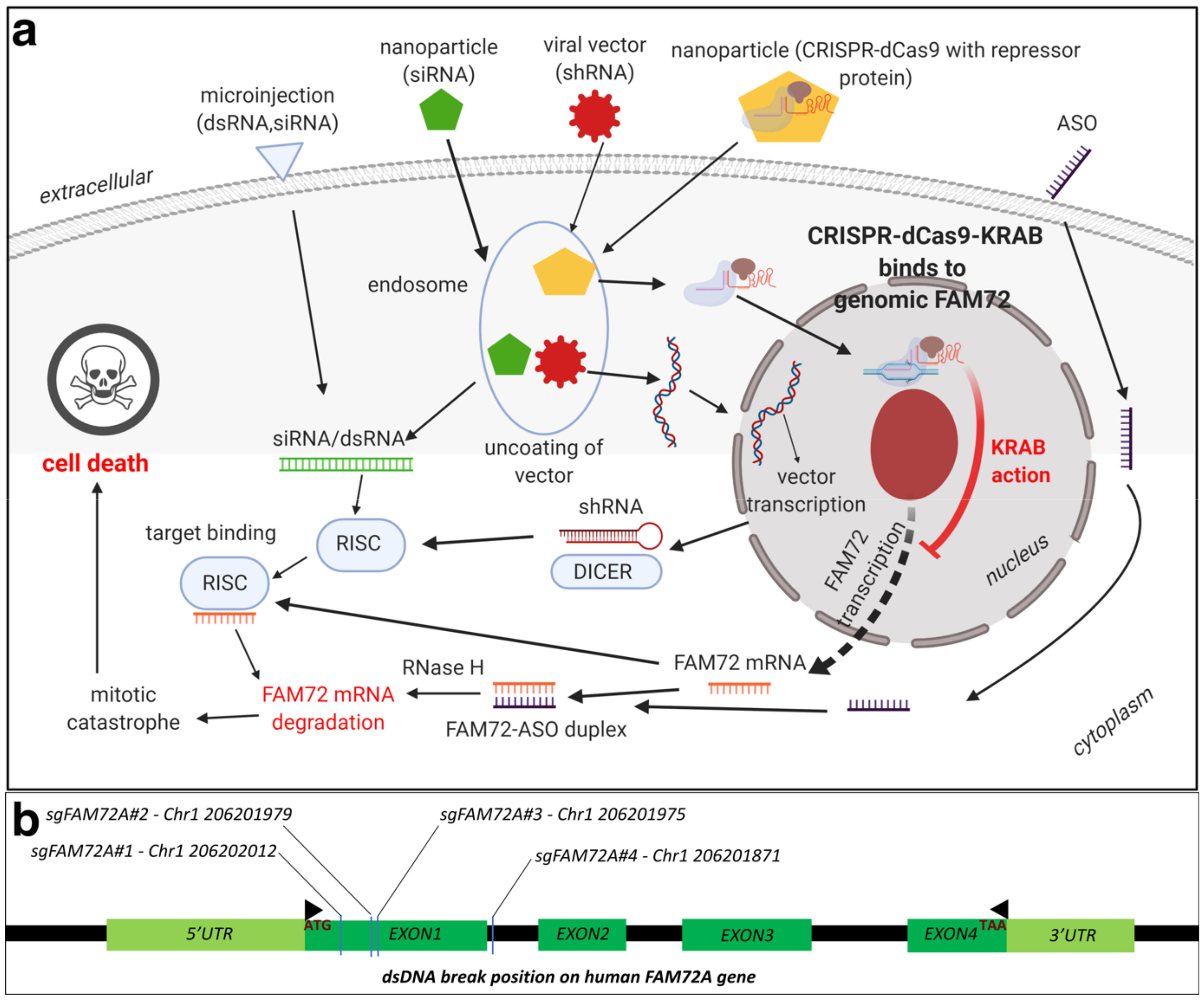
An alternative mechanism to knockout FAM72 in cancer tissues would be to use the clustered regularly interspersed short palindromic repeats (CRISPR)-CRISPR-associated protein (Cas) 9 gene editing tool. Briefly, CRISPR and Cas target foreign viral DNA as part of the adaptive immune system in bacteria [24]. A combination of trans-activating RNA (tracrRNA) and CRISPR targeting RNA (crRNA), together known as small guide RNA (sgRNA or sgFAM72-RNA), guide Cas proteins to the targeted foreign viral (or tumorigenic FAM72) DNA, which is then degraded [25]. The sg FAM72-RNA in combination with the Cas9 protein from Streptococcus pyogenes form the popular CRISPR-Cas9 gene editing tool [26][27][28]. A nuclease deficient Cas9 (dCas9) system combined with a transcriptional repressor protein such as the Kruppel-associated box (KRAB) [29][30] that target the transcription start site for FAM72 would be ideal to knockdown FAM72 in vivo at the site of the tumor [30][31][32][33][34]. Since FAM72 is overexpressed in non-neuronal cancer tissues, such a system would only affect the cancer tissues, leading to greater specificity. The delivery mechanism could be via lipid nanoparticles, similar to siRNA (Figure 3) [35].
2.3. Therapeutic Options against Tumorigenic FAM72: Chemotherapy
FAM72 and its paralogs could also be targeted via chemotherapy options using targeted drugs. We conducted an in silico binding study to predict potential ligand binding sites on FAM72A [36]. We found potential Zn2+ and Fe3+ binding sites along with possible binding for the organic compound RSM: (2s)-2-(acetylamino)-N-methyl-4-[(R)-methylsulfinyl] butanamide) [36].
Structure-based drug design (SBDD) is rapidly growing with the development of new technologies (e.g., high-throughput screening, molecular docking, pharmacophore mapping, quantitative structure-activity/property/toxicity relationship (QSAR/QSPR/QSTR), and virtual screening) to interpret, guide, and advance experimental biomedical research to achieve success in anti-cancer drug discovery [37][38][39][40][41]. SBDD methods analyze three-dimensional (3D) structures of macromolecule, typically of proteins or RNA, to identify key sites and interactions, which are important for their specific biological functions [38]. Understanding key sites and interactions can be used to design potential drug candidates that can interfere with essential interactions of the target protein and thus interrupt signaling pathways for survival and progression of cancer cells [38][42]. This requires knowledge of the 3D structure of the drug candidate and how its shape and charge cause it to interact with its biological target, ultimately revealing a therapeutic effect [38][43].
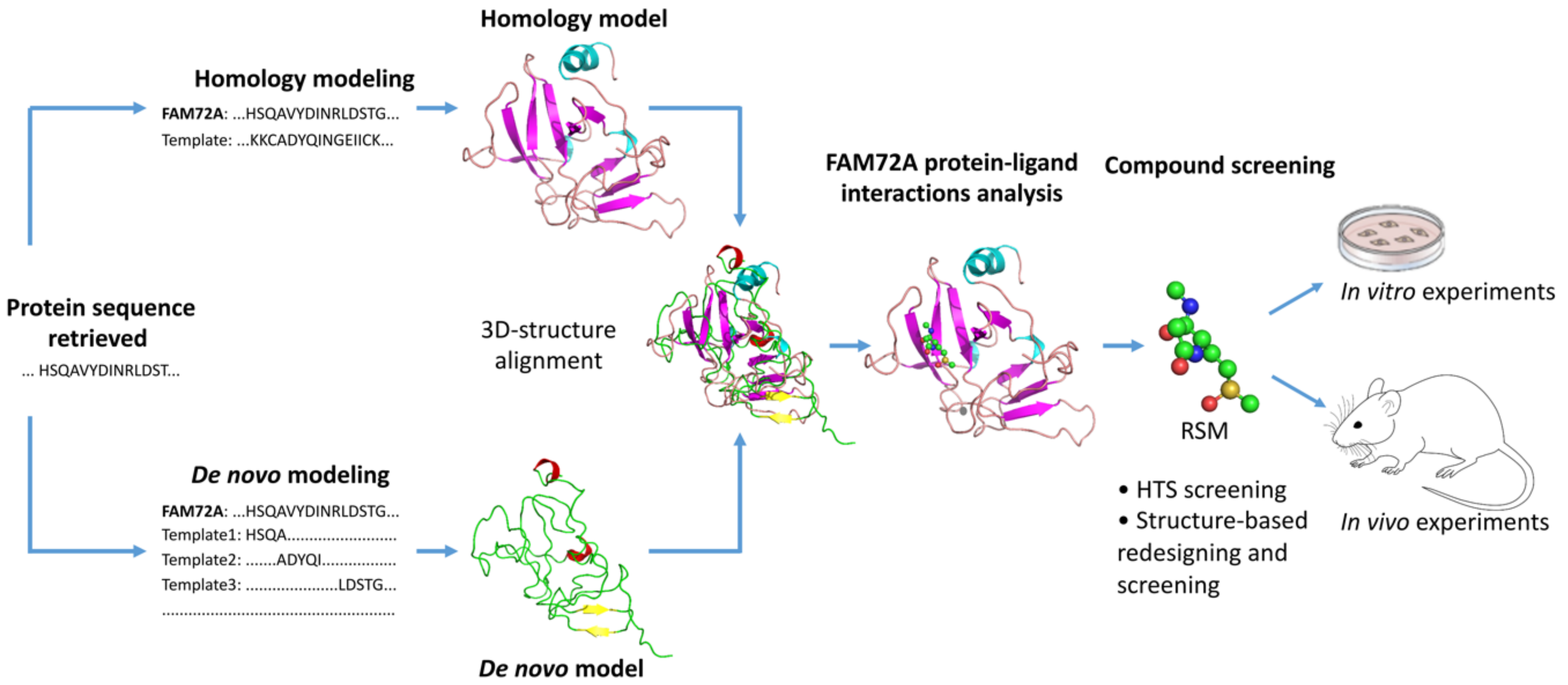
As discussed above in this review, increasing evidence indicates that FAM72 is a potential therapeutic target for the treatment of cancers [1][3][44][9], especially GBM [8] and adrenocortical carcinoma (ACC) [45]. In essence, 3D protein structures and understanding ligand–protein interactions of FAM72 represent the key and even obligatory steps in FAM72-targated drug design for the development of a useful treatment for GBM and ACC. There is an urgent need to advance the FAM72-targeted drug design process, and we employed a comprehensive in silico 3D protein determination strategy to determine the 3D protein structure of FAM72A and further identify potential ligand–protein interactions of FAM72A (Figure 4) [36]. An integrated approach combining homology modeling and de novo modeling was applied to obtain a reliable 3D protein structure of FAM72A [36]. In the homology modeling, a homologous template search was performed in various databases (e.g., National Center for Biotechnology Information-Protein Data Bank (NCBI-PDB), Phyre2, 3D-JIGSAW, Swiss Model, and RaptorX) [36]. Additionally, 3D FAM72A protein structure models were also obtained from Phyre2, 3DJIGSAW, Swiss Model, and RaptorX tools. Furthermore, an optimized prediction with the Modeller program [46][47][48] using templates, 1YQ3_D, 4OGC_A, 4OGE_A, 3GA3_A, 3MCA_B, 1I8D_B, 4M0M_A, 2FJA_A, and 3UK7_A (obtained from NCBI-PDB, Phyre2, 3D-JIGSAW, Swiss Model, and RaptorX) revealed that the monomeric 3D FAM72A protein structure, based on the 3GA3_A template, was the most reliable model in terms of stereochemical parameter evaluations (i.e., G-factor, Ramachandran plot analysis, and additional comparative iterative threading assembly refinement (I-TASSER) analysis) [36]. To this end, protein-ligand binding site prediction based on BioLiP protein function database screening (based on COACH, TM-SITE, S-SITE, COFACTOR, and ConCavity methods) [49][50] revealed that FAM72A is a Zn2+- or Fe3+-containing protein, which could potentially interact with the organic molecule RSM (Figure 4) [36]. Taken together, these data suggest a theoretical view of the 3D structure model of FAM72A and its ligand-binding sites [36]. In our view, these structural and protein–ligand interaction data provide a basis of FAM72A protein ligand-binding sites, which require further investigation using well-defined in vitro and in vivo experiments to confirm the therapeutic activity of the suggested compound as potential leads for drug discovery screenings for the treatment of FAM72A-driven cancers (e.g., GBM and ACC) [36].
This entry is from 10.3390/cancers13051025 and 10.3390/cancers13051025.
References
- Kutzner, A.; Pramanik, S.; Kim, P.S.; Heese, K. All-or-(N)One—An epistemological characterization of the human tumorigenic neuronal paralogous FAM72 gene loci. Genomics 2015, 106, 278–285.
- Ho, N.T.; Kim, P.S.; Kutzner, A.; Heese, K. Cognitive Functions: Human vs. Animal—4:1 Advantage |-FAM72-SRGAP2-|. J. Mol. Neurosci. 2017, 61, 603–606.
- Nehar, S.; Mishra, M.; Heese, K. Identification and characterisation of the novel amyloid-beta peptide-induced protein p17. FEBS Lett. 2009, 583, 3247–3253.
- Benayoun, B.A.; Pollina, E.A.; Ucar, D.; Mahmoudi, S.; Karra, K.; Wong, E.D.; Devarajan, K.; Daugherty, A.C.; Kundaje, A.B.; Mancini, E.; et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 2014, 158, 673–688.
- Ho, N.T.T.; Kutzner, A.; Heese, K. Brain plasticity, cognitive functions and neural stem cells: A pivotal role for the brain-specific neural master gene |-SRGAP2-FAM72-|. Biol. Chem. 2017, 399, 55–61.
- Dennis, M.Y.; Nuttle, X.; Sudmant, P.H.; Antonacci, F.; Graves, T.A.; Nefedov, M.; Rosenfeld, J.A.; Sajjadian, S.; Malig, M.; Kotkiewicz, H.; et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 2012, 149, 912–922.
- Geschwind, D.H.; Konopka, G. Neuroscience: Genes and human brain evolution. Nature 2012, 486, 481–482.
- Rahane, C.S.; Kutzner, A.; Heese, K. A cancer tissue-specific FAM72 expression profile defines a novel glioblastoma multiform (GBM) gene-mutation signature. J. Neurooncol. 2019, 141, 57–70.
- Heese, K. The protein p17 signaling pathways in cancer. Tumour Biol. 2013, 34, 4081–4087.
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856.
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552.
- Fang, L.; Cheng, Q.; Li, W.; Liu, J.; Li, L.; Xu, K.; Zheng, J. Antitumor activities of an oncolytic adenovirus equipped with a double siRNA targeting Ki67 and hTERT in renal cancer cells. Virus Res. 2014, 181, 61–71.
- Zhang, J.; Ding, M.; Xu, K.; Mao, L.; Zheng, J. shRNA-armed conditionally replicative adenoviruses: A promising approach for cancer therapy. Oncotarget 2016, 7, 29824–29834.
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293.
- Castanotto, D.; Stein, C.A. Antisense oligonucleotides in cancer. Curr. Opin. Oncol. 2014, 26, 584–589.
- Yang, C.; Zhang, J.; Ding, M.; Xu, K.; Li, L.; Mao, L.; Zheng, J. Ki67 targeted strategies for cancer therapy. Clin. Transl. Oncol. 2018, 20, 570–575.
- Kausch, I.; Lingnau, A.; Endl, E.; Sellmann, K.; Deinert, I.; Ratliff, T.L.; Jocham, D.; Sczakiel, G.; Gerdes, J.; Bohle, A. Antisense treatment against Ki-67 mRNA inhibits proliferation and tumor growth in vitro and in vivo. Int. J. Cancer 2003, 105, 710–716.
- D’Cruz, O.J.; Qazi, S.; Hwang, L.; Ng, K.; Trieu, V. Impact of targeting transforming growth factor beta-2 with antisense OT-101 on the cytokine and chemokine profile in patients with advanced pancreatic cancer. Onco Targets Ther. 2018, 11, 2779–2796.
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185.
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119.
- Odate, S.; Veschi, V.; Yan, S.; Lam, N.; Woessner, R.; Thiele, C.J. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin. Cancer Res. 2017, 23, 1771–1784.
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3331.
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446.
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823.
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Wang, X.; Ma, S.; Liu, Y.; Lu, W.; Sun, L.; Zhao, P.; Xia, Q. Transcriptional repression of endogenous genes in BmE cells using CRISPRi system. Insect Biochem. Mol. Biol. 2019, 111, 103172.
- MacLeod, R.S.; Cawley, K.M.; Gubrij, I.; Nookaew, I.; Onal, M.; O’Brien, C.A. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci. Rep. 2019, 9, 17312.
- Kampmann, M. CRISPRi and CRISPRa Screens in Mammalian Cells for Precision Biology and Medicine. ACS Chem. Biol. 2018, 13, 406–416.
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183.
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell. Biol. 2016, 17, 5–15.
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451.
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235.
- Pramanik, S.; Kutzner, A.; Heese, K. Lead discovery and in silico 3D structure modeling of tumorigenic FAM72A (p17). Tumour Biol. 2015, 36, 239–249.
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839.
- Yu, W.; MacKerell, A.D., Jr. Computer-Aided Drug Design Methods. In Methods in Molecular Biology; Springer: Berlin, Germany, 2017; Volume 1520, pp. 85–106.
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663.
- Pramanik, S.; Roy, K. Predictive modeling of chemical toxicity towards Pseudokirchneriella subcapitata using regression and classification based approaches. Ecotoxicol. Environ. Saf. 2014, 101, 184–190.
- Pramanik, S.; Roy, K. Exploring QSTR modeling and toxicophore mapping for identification of important molecular features contributing to the chemical toxicity in Escherichia coli. Toxicol. In Vitro 2014, 28, 265–272.
- van Montfort, R.L.; Workman, P. Structure-based design of molecular cancer therapeutics. Trends Biotechnol. 2009, 27, 315–328.
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22.
- Ho, N.T.T.; Kutzner, A.; Heese, K. A Novel Divergent Gene Transcription Paradigm-the Decisive, Brain-Specific, Neural |-Srgap2-Fam72a-| Master Gene Paradigm. Mol. Neurobiol. 2019, 56, 5891–5899.
- Rahane, C.S.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet. 2019, 230, 1–12.
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491.
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325.
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37.
- Yang, J.; Roy, A.; Zhang, Y. BioLiP: A semi-manually curated database for biologically relevant ligand-protein interactions. Nucl. Acids Res. 2013, 41, D1096–D1103.
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595.