Neurodegenerative diseases (NDs) are one of major public health problems and their impact is continuously growing. Curcumin has been proposed for the treatment of several of these pathologies, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) due to the ability of this molecule to reduce inflammation and aggregation of involved proteins.
- curcumin
- Alzheimer’s disease
- amyloid
- tau
1. Introduction
For last two centuries, natural occurring products have attracted the attention of many researchers due to their health benefits in the prevention and treatment of several diseases [1]. In 1815 Vogel isolated a yellow pigment, called curcumin, from the rhizome of Curcuma Longa, an East Indian plant [2]. Curcumin is the most abundant polyphenol and the most biologically active molecule found in the turmeric root; other minor components, known as curcuminoids, are demethoxycurcumin, bisdemethoxycurcumin, and cyclocurcumin [3]. Curcumin is one of the main elements of the Southeast Asian diet and it has been widely used for centuries as a traditional Indian and Asian medicine. After its first extraction, several studies showed that this polyphenolic molecule exhibits a broad spectrum of biological activities. Curcumin offers several health benefits, including anticancer [4], hypoglycemic activities [5], as well as the ability to be used as an analgesic, antiseptic or antimalarial [6]. In addition, curcumin has been shown to have anti-inflammatory [7], antioxidant [8,9][8][9], and antiamyloidogenic properties, which are relevant for the treatment of Alzheimer’s disease (AD), and related diseases [10,11][10][11].
Worldwide, 50 million people have dementia. Unfortunately, this number is expected to increase exponentially, affecting 152 million people by 2050 [12]. AD is the most prevalent progressive neurodegenerative disease associated with age and the most common form of dementia [13], contributing to 60–70% of cases. AD is characterized clinically by progressive loss of memory, language problems, social withdrawal, deterioration of executive functions and eventually death [14,15][14][15]. Histopathologically, as Alzheimer’s disease progresses, the brain shrinks dramatically and is characterized by cortex damage, and progressive degeneration of limbic and cortical brain structures, mainly in the temporal lobe [15]. This atrophy also affects the cortical association areas and the hippocampus, which is critical for the formation of new memories [16]. As a result of this pattern of cortical thinning, it is also possible to observe an enlargement of ventricles and a functional alteration of Wernicke’s and Broca’s areas [17]. A common characteristic of age-related neurodegenerative diseases, including AD, is the pathological accumulation of unfolded and aggregation-prone proteins in the brain, which are considered the major cause of synaptic loss and progressive neuronal death observed in these disorders [18]. The two major systems involved in proteostasis maintenance are the autophagy-lysosomal system and the ubiquitin proteasome system [19]. However, these two systems have been found to be impaired in many neurodegenerative diseases, including AD. Therefore, the failure of these systems in maintaining proteostasis may also contribute to the pathological aggregation of proteins as well as formation of insoluble and fibrillar amyloid inclusions [20].
The major neuropathological features of AD are synaptic and neuronal degeneration and the presence of amyloid plaques and neurofibrillary tangles (NFTs).
Neuritic plaques are polymorphous aggregates made up of the amyloid Aβ peptide (Aβ) aggregates. The ≈4 kDa Aβ fragment originates from the transmembrane amyloid precursor protein (APP) by concerted proteolytic cleavage of β- and γ-secretase [21]. Monomeric Aβ1-40 (Aβ40) and Aβ1-42 (Aβ42) species can aggregate to form Aβ oligomers that can further aggregate and assembly into amyloid fibrils [22]. A growing body of evidence suggest that the oligomeric/prefibrillar Aβ peptide is the neurotoxic species that trigger the amyloid cascade, leading to the damage and eventual death of neurons associated with AD [23,24,25][23][24][25]. On the other hand, NFTs are intracellular inclusions of hyperphosphorylated tau, a microtubule associated protein. In its native state tau is a monomeric protein [26]. Tau is a natively unfolded protein involved in microtubule stabilization and axonal transport. However, under pathological conditions, tau can undergo abnormal post-translational modifications, including phosphorylation or acetylation [27,28][27][28]. As result of these modifications, tau detaches from the microtubules causing their disassembly, cytoskeletal instability, and axonal transport perturbation [29,30][29][30]. Unbound tau can self-aggregate forming soluble tau oligomers that assemble into paired helical filaments (PHFs) [31,32,33][31][32][33]. The PHFs mature into fibrils that constitute the intracellular NFTs, observed in the brain of AD patients [27]. Increasing evidence suggests that synaptic dysfunction and neuronal loss precede the formation of NFTs [34[34][35][36][37][38][39],35,36,37,38,39], indicating that the smaller and prefibrillar aggregates, tau oligomers, may be responsible for the toxic effects during the early stage of AD and other tauopathies [40,41][40][41]. Therefore, tau oligomers are considered to be highly toxic and to seed tau misfolding, thus propagating the pathology seen across different neurodegenerative diseases [38,42][38][42].
Despite the many efforts made to develop new treatments and therapeutic approaches to prevent the onset of the disease and to reverse the disease process, to date, there are no effective therapeutics. Nowadays, the therapeutic strategies available are only symptomatic treatments that counterbalance neurotransmitter disturbance, thus ameliorating a few of the clinical symptoms associated with the disease. The established treatments available are acetylcholinesterase inhibitors (e.g., Donepezil or Tacrine), antagonists of glutamate NMDA receptor (e.g., Memantine), agonist of nicotinic or muscarinic receptors, antioxidants and anti-inflammatory agents [43,44][43][44].
Growing evidence demonstrates a protective effect of curcumin against Aβ plaque formation; however, the mechanism of action is not yet fully clarified. Some studies have classified curcumin as an inhibitor of Aβ aggregation, others as disaggregating and destabilizing of amyloid fibrils [45]. In addition, curcumin has been shown to hamper Aβ oligomerization but not its fibrillization [46]. Recently, curcumin has been shown to attenuate amyloid-β aggregate-associated neurotoxicity by promoting the formation of “off-pathway” nontoxic soluble oligomers and prefibrillar proteins [47].
Curcumin has also been shown to exert a neuroprotective role by inhibiting tau aggregation. Indeed, curcumin has been shown to inhibit tau oligomerization, disintegrate preformed tau oligomers, inhibit β-sheet formation, and disaggregate tau filaments [48,49][48][49]. In addition, in vitro studies have shown that curcumin prevents the aggregation of other amyloidogenic protein, including α-synuclein (α-syn), which is a presynaptic protein involved in PD. PD is a debilitating neurodegenerative disorder characterized by the gradual loss of dopaminergic neurons in the substantia nigra pars compacta and clinically characterized as movement disorder. α-syn accumulates abnormally and aggregates in the cytosol as Lewy bodies and in the neuronal processes as Lewy neurites [50]. Several studies have showed that curcumin inhibits α–syn aggregation and reduces α-syn-induced cytotoxicity [51,52][51][52].
The neuroprotective effect of curcumin is certainly due to its ability to modulate the aggregation pathways and toxicity of amyloidogenic proteins and mitigate inflammation and oxidative stress, known to be key factors in the progression of neurodegenerative disorders [53,54][53][54].
2. Physicochemical Characteristics of Curcumin
Due to the relevant biological and health benefits of curcumin, several chemists proposed a potential structure of curcumin. In 1913 Lampe et al. synthesized curcumin for the first time [55]. A general procedure for the synthesis of curcumin with a higher yield was later reported by Pabon [56]. In this reaction scheme 2,4-diketones, such as acetyl acetone, reacts with conveniently substituted aromatic aldehydes, particularly vanillin aldehyde, to synthesize curcumin. To prevent a Knoevenagel condensation due to the high acidity of the α-methylene group, the reaction is carried out in the presence of boron oxide as a complexing agent for the dienolate group. In this way, the condensation reaction involves terminal alkyl groups of di-ketone and primary or secondary amines, usually n-butylamine, are used to deprotonate these groups. Alkyl borates act as drying agents to remove the water formed by condensation reaction between boron complex and aromatic aldehyde. In the final step, boron complex gives the final product in acidic conditions. The reaction is refluxed, using aprotic solvent such as ethyl acetate (Scheme 1).
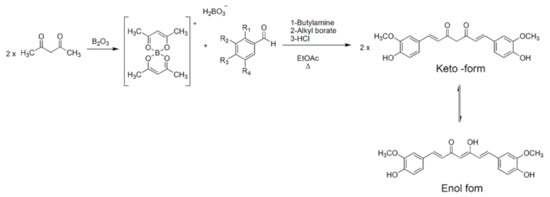
Scheme 1. Synthesis of curcumin with Pabon’s method.
Several research groups follow the general method proposed by Pabon with slight modifications. For example, boron oxide has been replaced with boric acid with a lower yield [57,58][57][58]. An alternative procedure, reported by Rao et al., replaced boron oxide with borontrifluoride to obtain curcuminoid difluoroboronites that can be then hydrolyzed using aqueous methanol at pH 5.8 to get curcuminoid compounds [59]. To synthesize polyhydroxy curcuminoids, it is necessary to protect the hydroxyls groups on the starting benzaldehyde. These groups were protected as ethers and deprotected using aluminum chloride [60]. Curcumin is a low molecular mass polyphenolic compound (368.38 g/moL) with a melting point of 183 °C [61]. The IUPAC name of curcumin is 1,7-bis(4-hydroxy-3-methoxy-phenyl)-1,6-heptadiene-3,5-dione and is also known as diferuloyl methane. Curcumin is a hydrophobic molecule with a log p value of 3.29. It is insoluble in water and soluble in polar organic solvent, like methanol, ethanol, dimethylsulfoxide, dimethylformamide, or ethyl acetate. It is partially soluble in hexane or cyclohexane [62].
Curcumin is a symmetrical molecule composed of two aromatic rings substituted with o-methoxy phenolic groups and a β-diketone moiety as a central linker. The heptadienone linkage exhibits keto-enol tautomerism (Scheme 1) that influences physicochemical and antioxidant properties of curcumin [63,64][63][64]. Curcumin is present in its bis-keto form in acidic and neutral pH conditions (pH 3–7) due to the presence of an acid proton linked to a highly activated carbon between the two aromatic rings. Conversely, under basic conditions (pH > 8), the enol form predominately and curcumin acts as an electron donor. Indeed, the antioxidant activity of curcumin is attributed to its enolic form [65]. X-ray crystallography studies confirmed that the enol form has a lower energy as compared to the diketone tautomer and it is the exclusive form in solution [66]. Moreover, keto-enol tautomer can exist as syn and anti isomers with the syn-enol form being more stable. In the syn form the two methoxy groups are on the same side with respect to keto-enol and hydroxy groups. Thus, it is possible to identify a polar surface with either a phenolic or enol group and a nonpolar area with methoxy groups [67].
Curcumin, as well as other polyphenolic compounds, displays a strong absorption in the visible region with a maximum absorption around 410–430 nm and another band with maximum absorption at 265 nm. In the presence of nonpolar solvents, including hexane or cyclohexane, a blue-shift of the absorption spectrum is observed. Conversely, in polar solvents, such as methanol or DMSO, the peak is shifted towards the lower frequencies [65]. These observations can be justified by the shift of the keto-enol tautomerism towards the enol form in a polar solvent or towards the bis-keto form in nonpolar solvent. The enol form exhibits a larger electronic delocalization and, therefore, a red-shifted absorption peak is observed [68].
43. Relationship between Structural Properties and Biological Activity of Curcumin Derivatives
The synthesis of novel curcumin derivatives represents an effective alternative to obtain curcumin analogs with a better solubility in biofluid in an effort to improve the pharmacokinetic profile of curcumin and its biological activity [48,86,87,88][48][69][70][71]. As mentioned above, curcumin acts as a neuroprotective agent blocking multiple mechanisms involved in neurodegeneration by interfering with the accumulation of misfolded aggregate proteins, including Aβ and tau, inflammation and oxidative stress. The modulation of each of these pathological pathways requires distinct structural feature of curcumin. Therefore, comprehensive structure–activity studies are extremely important to identify novel curcumin derivatives as potential therapeutic agents for neurodegenerative diseases.
In recent years, researchers have synthesized several compounds able to block Aβ fibrillogenesis. In this process, Aβ monomers aggregate to form oligomers, which then assemble to form insoluble aggregates [89][72]. This transformation is characterized by a structural transition from α-helix to β-sheet structure [90][73]. It is known that the short Aβ fragment, KLVFF (Aβ16-20) binds to full length Aβ and it is important for amyloid fibril formation [91][74]. A shared model hypothesizes that phenylalanine residue in the KLVFF sequence of Aβ peptides interact through Π-Π interactions during Aβ aggregation. Small molecules, such as curcumin, are able to block or break these interactions and could be valid candidates to revert amyloid formation [92][75].
Reinke and Gestwicki have created a library of compounds resembling curcumin structure to investigate and evaluate the effect of the three main features of curcumin on inhibition of amyloid aggregation [93][76]. The structural features contributing to the inhibitory potency of curcumin are the two aromatic rings, the substitution pattern of these phenyl groups, and the length and flexibility of the central linker region. To perform structural considerations and better understand which feature is critical for the inhibition of Aβ aggregation, Reinke and Gestwicki synthesized curcumin analogs by modifying only one structural feature at the time and retaining the other two. As a result of their studies, they found that compounds lacking one aromatic group are less active than curcumin, suggesting that both aromatic rings are essential to interact through hydrophobic interactions and hydrogen bonding with phenylalanine residue of Aβ monomers and inhibit amyloid formation [94][77]. Furthermore, hydroxyl substitution on the aromatic end group or other polar functional substituents are required for the inhibiting activity. In addition, Reinke and Gestwicki showed that both length and flexibility of the central linker region are key factors to take into consideration in the design of new Aβ aggregation inhibitors. Indeed, the inhibiting activity is negatively affected when the central linker region is too long, too short, or too flexible. The optimal length of the central linker is 8–16 Å and no more than one or two rotating sp3-hybridized carbons are required for an ideal flexibility.
It has been shown that the homeostasis of metal ions is critical for maintaining normal physiological functions. Some ions such as Al, Fe, Cu, and Zn have been observed in the brain of AD patients [95][78]. Their imbalance in the brain is closely related to the Aβ deposition and tau accumulation, suggesting that they play a role in the degenerative process of AD. The histidine residues of Aβ peptides (H13/H14) are good coordination sites for metal ions [96][79]. Curcumin can interact with metal ions forming strong complexes. Indeed, the α,β-unsaturated β-diketo moiety of curcumin has shown excellent chelating properties. Many studies reported the synthesis of stable metal-curcumin complex with a stoichiometry 2:1 (ligand:metal) [62]. Curcumin-metal complexes decrease Aβ plaques as well as suppress inflammatory processes by preventing metal induction of nuclear factor kappa B, NF-kB [97,98][80][81]; however, metal chelators can disrupt the normal brain homeostasis. Zhang et al. designed a novel curcumin derivative, named CRANAD-17, as a chelating agent to attenuate Aβ crosslinking induced by Cu [99][82].
Some curcumin derivatives exert their neuroprotective effects by promoting phagocytosis of Aβ fibrils. For example, it was demonstrated by Fiala et al. that bisdemethoxycurcumin (BDC) enhances macrophage-activated Aβ clearance and reduces the inflammation state [100][83]. Recently, Gagliardi et al. showed that the treatment of BDC derivatives in a human monocytic cell line, mimicking the peripheral blood mononuclear cells of AD, revealed overexpression of genes essential for macrophage function, including mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase and vitamin D receptor. BDC also showed a protective anti-inflammatory effect through downregulation of NF-kB and β-site APP cleaving enzyme 1 (BACE1) genes [101][84]. BACE 1 protein and γ-secretase enzyme cleaves APP to generate Aβ peptides, with the β-cleavage being the rate-limiting step of this sequential proteolytic pathway [102][85]. Inhibitors of BACE1 activity are, therefore, considered as a possible therapeutic approach and many BACE1 inhibitors have been synthesized. Two critical curcumin structural features associated with BACE 1 inhibitory activity are the phenolic rings and the unsaturated alkenyl linker between the aromatic rings. Derivatives with multiple hydroxy groups have been found to be more active than compounds with nonsubstituted phenyl groups or substituted with methoxy groups or halogen. When researchers replaced the phenol group with indole or a pyrrole ring, they produced more active compounds. These data suggest that inhibitor molecules interact with BACE 1 through hydrogen bonds [103][86]. Reduction products are not active against BACE 1. A planar structure is required to maintain the inhibitory activity and sp2 carbons give the optimal rigidity to the molecule. The substitution of 1,3-dicarbonyl moiety of curcumin by isosteric heterocycles, isoxazole, and pyrazole, resulted in the formation of potent inhibitors of γ-secretase enzyme [104][87].
Up to this point of our discussion, we have analyzed the structural aspects of curcuminoids that have been found to be important for their anti-Aβ aggregation activity. Curcumin and its derivatives also exert a protective role against misfolded tau aggregates. Unlike Aβ, tau lacks in hydrophobic residues and therefore its aggregation process does not lead to the formation of Π-Π interactions. However, under particular circumstances, the small degree of hydrophobicity, compared to other proteins, is sufficient to drive tau aggregation [105][88]. Tau aggregation inhibitors interact with tau by electrostatic interaction and hydrogen bonding. A recent study suggested two ruthenium-curcumin-bipyridine/phenanthroline complexes as inhibitors of tau aggregation. In these complexes, the metal ion is bound to the enol group of curcumin and to the nitrogen atoms of the ancillary ligands, bipyridine or phenanthroline. Curcumin inhibits aggregation at the nucleation stage while the positive charged ruthenium complex inhibits the elongation phase, reducing longer fibrils formation [106][89].
Oxidative damage plays an important role in neurodegeneration, therefore, to treat neurodegenerative disorders another pharmacological approach is to develop antioxidant compounds. The antioxidant property of curcumin is due to the abstractable phenolic hydrogen [107][90]. This group can reduce, for example, superoxide radicals to generate less reactive phenoxyl radicals that are resonance stabilized [108][91]. Ferrari et al. developed curcumin analogs substituted on the central carbon of the heptadienone linker and demonstrated that these complexes exhibit good metal chelating properties. However, these compounds showed less scavenging activity because of the presence of the substituent on central linker shifts the keto-enol tautomerism towards the di-keto form with less stabilization of phenoxyl radical [109][92].
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661.
- Vogel, A.; Pelletier, J. Examen chimique de la racine de Curcuma. J. Pharm. 1815, 1, 289–300.
- Ravindran, P.N.; Babu, K.N.; Sivaraman, K. Turmeric: The Genus Curcuma; Taylor & Francis: Boca Raton, FL, USA, 2007.
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10, 12.
- Nishiyama, T.; Mae, T.; Kishida, H.; Tsukagawa, M.; Mimaki, Y.; Kuroda, M.; Sashida, Y.; Takahashi, K.; Kawada, T.; Nakagawa, K.; et al. Curcuminoids and sesquiterpenoids in turmeric (Curcuma longa L.) suppress an increase in blood glucose level in type 2 diabetic KK-Ay mice. J. Agric. Food Chem. 2005, 53, 959–963.
- Prasad, S.; Aggarwal, B.B. Turmeric, the Golden Spice: From Traditional Medicine to Modern Medicine. In Herbal Medicine: Biomolecular and Clinical Aspects; Benzie, I.F.F., Wachtel-Galor, S., Eds.; Taylor and Francis Group, LLC.: Boca Raton, FL, USA, 2011.
- Hatami, M.; Abdolahi, M.; Soveyd, N.; Djalali, M.; Togha, M.; Honarvar, N.M. Molecular Mechanisms of Curcumin in Neuroinflammatory Disorders: A Mini Review of Current Evidences. Endocr. Metab. Immune Disord. Drug Targets 2019, 19, 247–258.
- Sharma, O.P. Antioxidant activity of curcumin and related compounds. Biochem. Pharmacol. 1976, 25, 1811–1812.
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Rafiee, R.; Sathyapalan, T.; Sahebkar, A. Effects of curcumin on mitochondria in neurodegenerative diseases. Biofactors (Oxf. Engl.) 2020, 46, 5–20.
- Lakey-Beitia, J.; Berrocal, R.; Rao, K.S.; Durant, A.A. Polyphenols as therapeutic molecules in Alzheimer’s disease through modulating amyloid pathways. Mol. Neurobiol. 2015, 51, 466–479.
- Maiti, P.; Dunbar, G.L. Use of Curcumin, a Natural Polyphenol for Targeting Molecular Pathways in Treating Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1637.
- World Health Organization. Dementia. 21 September 2020. Available online: (accessed on 9 February 2021).
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharm. Sci. 1991, 12, 383–388.
- Citron, M. Alzheimer’s disease: Treatments in discovery and development. Nat. Neurosci. 2002, 5, 1055–1057.
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148.
- Jahn, H. Memory loss in Alzheimer’s disease. Dialogues Clin. Neurosci. 2013, 15, 445–454.
- Mesulam, M.M.; Thompson, C.K.; Weintraub, S.; Rogalski, E.J. The Wernicke conundrum and the anatomy of language comprehension in primary progressive aphasia. Brain A J. Neurol. 2015, 138, 2423–2437.
- Walker, L.C.; LeVine, H. The cerebral proteopathies. Mol. Neurobiol. 2000, 21, 83–95.
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta 2008, 1782, 691–699.
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6.
- Selkoe, D.J. Alzheimer’s disease: A central role for amyloid. J. Neuropathol. Exp. Neurol. 1994, 53, 438–447.
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68.
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, S74–S78.
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489.
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357.
- Naseri, N.N.; Wang, H.; Guo, J.; Sharma, M.; Luo, W. The complexity of tau in Alzheimer’s disease. Neurosci. Lett. 2019, 705, 183–194.
- Alonso, A.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928.
- Cook, C.; Stankowski, J.N.; Carlomagno, Y.; Stetler, C.; Petrucelli, L. Acetylation: A new key to unlock tau’s role in neurodegeneration. Alzheimer’s Res. Ther. 2014, 2, 29.
- Alonso, A.D.; Cohen, L.S.; Corbo, C.; Morozova, V.; ElIdrissi, A.; Phillips, G.; Kleiman, F.E. Hyperphosphorylation of Tau Associates with Changes in Its Function Beyond Microtubule Stability. Front. Cell. Neurosci. 2018, 12, 338.
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506.
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 2408–2421.
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134.
- Belostozky, A.; Richman, M.; Lisniansky, E.; Tovchygrechko, A.; Chill, J.H.; Rahimipour, S. Inhibition of tau-derived hexapeptide aggregation and toxicity by a self-assembled cyclic d,l-alpha-peptide conformational inhibitor. Chem. Commun. (Camb. Engl.) 2018, 54, 5980–5983.
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24.
- Terry, R.D. Do neuronal inclusions kill the cell? J. Neural Transm. Suppl. 2000, 59, 91–93.
- Van De Nes, J.A.; Nafe, R.; Schlote, W. Non-tau based neuronal degeneration in Alzheimer’s disease -- an immunocytochemical and quantitative study in the supragranular layers of the middle temporal neocortex. Brain Res. 2008, 1213, 152–165.
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res 2006, 54, 197–201.
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012.
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076.
- Gerson, J.E.; Castillo-Carranza, D.L.; Kayed, R. Advances in therapeutics for neurodegenerative tauopathies: Moving toward the specific targeting of the most toxic tau species. ACS Chem. Neurosci. 2014, 5, 752–769.
- Gerson, J.E.; Kayed, R. Formation and propagation of tau oligomeric seeds. Front. Neurol. 2013, 4, 93.
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 14234–14250.
- Mayeux, R.; Sano, M. Treatment of Alzheimer’s disease. N. Engl. J. Med. 1999, 341, 1670–1679.
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341.
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901.
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C.G. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324.
- Thapa, A.; Jett, S.D.; Chi, E.Y. Curcumin Attenuates Amyloid-β Aggregate Toxicity and Modulates Amyloid-β Aggregation Pathway. ACS Chem. Neurosci. 2016, 7, 56–68.
- Ma, Z.; Wang, N.; He, H.; Tang, X. Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application. J. Control. Release Off. J. Control. Release Soc. 2019, 316, 359–380.
- Rane, J.S.; Bhaumik, P.; Panda, D. Curcumin Inhibits Tau Aggregation and Disintegrates Preformed Tau Filaments in vitro. J. Alzheimer’s Dis. JAD 2017, 60, 999–1014.
- Pandey, N.; Strider, J.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathol. 2008, 115, 479–489.
- Wang, M.S.; Boddapati, S.; Emadi, S.; Sierks, M.R. Curcumin reduces α-synuclein induced cytotoxicity in Parkinson’s disease cell model. BMC Neurosci. 2010, 11, 57.
- Liu, Z.; Yu, Y.; Li, X.; Ross, C.A.; Smith, W.W. Curcumin protects against A53T alpha-synuclein-induced toxicity in a PC12 inducible cell model for Parkinsonism. Pharmacol. Res. 2011, 63, 439–444.
- Kim, G.Y.; Kim, K.H.; Lee, S.H.; Yoon, M.S.; Lee, H.J.; Moon, D.O.; Lee, C.M.; Ahn, S.C.; Park, Y.C.; Park, Y.M. Curcumin inhibits immunostimulatory function of dendritic cells: MAPKs and translocation of NF-kappa B as potential targets. J. Immunol. 2005, 174, 8116–8124.
- Baum, L.; Ng, A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimer’s Dis. JAD 2004, 6, 367–377.
- Lampe, V.; Milobedzka, J. Studien über Curcumin. Ber. Der Dtsch. Chem. Ges. 2006, 46, 2235–2240.
- Pabon, H.J.J. A synthesis of curcumin and related compounds. Recueil Travaux Chimiques Pays-Bas 1964, 83, 379–386.
- Krackov, M.H.; Bellis, H.E. Process for the Synthesis of Curcumin Related Compounds. US Patent 5,679,864, 21 October 1997.
- Babu, K.V.D.; Rajasekharan, K.N. Simplified condition for synthesis of Curcumin I and other curcuminoids. Org. Prep. Proced. Int. 1994, 26, 674–677.
- Rao, E.V.; Sudheer, P. Revisiting curcumin chemistry part I: A new strategy for the synthesis of curcuminoids. Indian J. Pharm. Sci. 2011, 73, 262–270.
- Venkateswarlu, S.; Ramachandra, M.S.; Subbaraju, G.V. Synthesis and biological evaluation of polyhydroxycurcuminoids. Bioorg. Med. Chem. 2005, 13, 6374–6380.
- Buadonpri, W.; Wichitnithad, W.; Rojsitthisak, P.; Towiwat, P. Synthetic Curcumin Inhibits Carrageenan-Induced Paw Edema in Rats. J. Health Res. 2018, 23, 11–16.
- Priyadarsini, K.I. The chemistry of curcumin: From extraction to therapeutic agent. Molecules 2014, 19, 20091–20112.
- Jovanovic, S.V.; Steenken, S.; Boone, C.W.; Simic, M.G. H-Atom Transfer Is A Preferred Antioxidant Mechanism of Curcumin. J. Am. Chem. Soc. 1999, 121, 9677–9681.
- Basnet, P.; Skalko-Basnet, N. Curcumin: An anti-inflammatory molecule from a curry spice on the path to cancer treatment. Molecules 2011, 16, 4567–4598.
- Lee, W.-H.; Loo, C.-Y.; Bebawy, M.; Luk, F.; Mason, R.S.; Rohanizadeh, R. Curcumin and its derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr. Neuropharmacol. 2013, 11, 338–378.
- Parimita, S.P.; Ramshankar, Y.V.; Suresh, S.; Guru Row, T.N. Redetermination of curcumin: (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,4,6-trien-3-one. Acta Crystallogr. Sect. E 2007, 63, o860–o862.
- Balasubramanian, K. Molecular Orbital Basis for Yellow Curry Spice Curcumin’s Prevention of Alzheimer’s Disease. J. Agric. Food Chem. 2006, 54, 3512–3520.
- Chignell, C.F.; Bilski, P.; Reszka, K.J.; Motten, A.G.; Sik, R.H.; Dahl, T.A. Spectral and photochemical properties of curcumin. Photochem. Photobiol. 1994, 59, 295–302.
- Farkhondeh, T.; Samarghandian, S.; Pourbagher-Shahri, A.M.; Sedaghat, M. The impact of curcumin and its modified formulations on Alzheimer’s disease. J. Cell. Physiol. 2019, 234, 16953–16965.
- Lo Cascio, F.; Puangmalai, N.; Ellsworth, A.; Bucchieri, F.; Pace, A.; Palumbo Piccionello, A.; Kayed, R. Toxic Tau Oligomers Modulated by Novel Curcumin Derivatives. Sci. Rep. 2019, 9, 19011.
- Pithadia, A.S.; Bhunia, A.; Sribalan, R.; Padmini, V.; Fierke, C.A.; Ramamoorthy, A. Influence of a curcumin derivative on hIAPP aggregation in the absence and presence of lipid membranes. Chem. Commun. (Camb. Engl.) 2016, 52, 942–945.
- Murphy, R.M. Kinetics of amyloid formation and membrane interaction with amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1923–1934.
- Fändrich, M.; Schmidt, M.; Grigorieff, N. Recent progress in understanding Alzheimer’s β-amyloid structures. Trends Biochem. Sci. 2011, 36, 338–345.
- Tjernberg, L.O.; Näslund, J.; Lindqvist, F.; Johansson, J.; Karlström, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548.
- Belluti, F.; Rampa, A.; Gobbi, S.; Bisi, A. Small-molecule inhibitors/modulators of amyloid-β peptide aggregation and toxicity for the treatment of Alzheimer’s disease: A patent review (2010–2012). Expert Opin. Ther. Pat. 2013, 23, 581–596.
- Reinke, A.A.; Gestwicki, J.E. Structure-activity relationships of amyloid beta-aggregation inhibitors based on curcumin: Influence of linker length and flexibility. Chem. Biol. Drug Des. 2007, 70, 206–215.
- Buchete, N.-V.; Hummer, G. Structure and dynamics of parallel beta-sheets, hydrophobic core, and loops in Alzheimer’s A beta fibrils. Biophys. J. 2007, 92, 3032–3039.
- Miller, L.M.; Wang, Q.; Telivala, T.P.; Smith, R.J.; Lanzirotti, A.; Miklossy, J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J. Struct. Biol. 2006, 155, 30–37.
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Aβ peptides. JBIC J. Biol. Inorg. Chem. 2004, 9, 954–960.
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035.
- Aggarwal, B.B.; Harikumar, K.B. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int. J. Biochem. Cell Biol. 2009, 41, 40–59.
- Zhang, X.; Tian, Y.; Li, Z.; Tian, X.; Sun, H.; Liu, H.; Moore, A.; Ran, C. Design and Synthesis of Curcumin Analogues for in Vivo Fluorescence Imaging and Inhibiting Copper-Induced Cross-Linking of Amyloid Beta Species in Alzheimer’s Disease. J. Am. Chem. Soc. 2013, 135, 16397–16409.
- Fiala, M.; Liu, P.T.; Espinosa-Jeffrey, A.; Rosenthal, M.J.; Bernard, G.; Ringman, J.M.; Sayre, J.; Zhang, L.; Zaghi, J.; Dejbakhsh, S.; et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc. Natl. Acad. Sci. USA 2007, 104, 12849–12854.
- Gagliardi, S.; Franco, V.; Sorrentino, S.; Zucca, S.; Pandini, C.; Rota, P.; Bernuzzi, S.; Costa, A.; Sinforiani, E.; Pansarasa, O.; et al. Curcumin and Novel Synthetic Analogs in Cell-Based Studies of Alzheimer’s Disease. Front. Pharmacol. 2018, 9, 1404.
- Das, U.; Wang, L.; Ganguly, A.; Saikia, J.M.; Wagner, S.L.; Koo, E.H.; Roy, S. Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat. Neurosci. 2016, 19, 55–64.
- Konno, H.; Endo, H.; Ise, S.; Miyazaki, K.; Aoki, H.; Sanjoh, A.; Kobayashi, K.; Hattori, Y.; Akaji, K. Synthesis and evaluation of curcumin derivatives toward an inhibitor of beta-site amyloid precursor protein cleaving enzyme 1. Bioorganic Med. Chem. Lett. 2014, 24, 685–690.
- Narlawar, R.; Pickhardt, M.; Leuchtenberger, S.; Baumann, K.; Krause, S.; Dyrks, T.; Weggen, S.; Mandelkow, E.; Schmidt, B. Curcumin-derived pyrazoles and isoxazoles: Swiss army knives or blunt tools for Alzheimer’s disease? ChemMedChem 2008, 3, 165–172.
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262.
- Liu, W.; Hu, X.; Zhou, L.; Tu, Y.; Shi, S.; Yao, T. Orientation-Inspired Perspective on Molecular Inhibitor of Tau Aggregation by Curcumin Conjugated with Ruthenium(II) Complex Scaffold. J. Phys. Chem. B 2020, 124, 2343–2353.
- Priyadarsini, K.I.; Maity, D.K.; Naik, G.H.; Kumar, M.S.; Unnikrishnan, M.K.; Satav, J.G.; Mohan, H. Role of phenolic O-H and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic. Biol. Med. 2003, 35, 475–484.
- Mishra, B.; Priyadarsini, K.I.; Bhide, M.K.; Kadam, R.M.; Mohan, H. Reactions of superoxide radicals with curcumin: Probable mechanisms by optical spectroscopy and EPR. Free Radic. Res. 2004, 38, 355–362.
- Ferrari, E.; Benassi, R.; Saladini, M.; Orteca, G.; Gazova, Z.; Siposova, K. In vitro study on potential pharmacological activity of curcumin analogues and their copper complexes. Chem. Biol. Drug Des. 2017, 89, 411–419.