A family of adiponectin paralogs designated as C1q complement/tumor necrosis factor (TNF)-associated proteins (CTRPs) has been found to play a role in the development of CVD. CTRPs, which are comprised of 15 members, CTRP1 to CTRP15, are secreted from different organs/tissues and exhibit diverse functions, have attracted increasing attention because of their roles in maintaining inner homeostasis by regulating metabolism, inflammation, and immune surveillance.
- CTRPs
- cardiovascular disease
- COVID-19
- obesity
1. Introduction
The CTRP superfamily, originally introduced by Harvey Lodish and colleagues, describes a new family of secreted proteins [5,6][1][2]. The CTRP superfamily is a paralog of adiponectin (APN), composed of CTRP1–CTRP15, which share a common structural domain with APN [5][1]. CTRPs are composed of four distinct domains, comprising an N-terminal signal peptide, a short variable domain, a collagen-like domain, and a C-terminal C1q-like globular domain. C1q forms trimers composed of A, B, and C chains [7][3]. (Figure 1) The globular domain is important for its biological function. The C-terminal region of the tumor necrosis factor (TNF) homology domain (THD), which is similar to that of the globular C1q (gC1q) domain, is a typical feature of members of the TNF family. The C1q and TNF family proteins have similar gene structures: their gC1q or THD domains are each encoded within one exon, while introns are restricted to respective stalk regions [7,8][3][4]. CTRPs are secreted by different viscera and tissues, including the adipose tissue, heart, and liver [9][5]. These adipokines play important roles in obesity, diabetes, and cardiovascular disease. According to current research, CTRP1, CTRP3, CTRP5, CTRP6, CTRP9, CTRP12, CTRP13, and CTRP15 are related to CVD. These proteins influence the progression of CVD by regulating inflammatory responses, endothelial function, metabolic dysfunction, myocardial cell apoptosis, and fibrosis [10,11][6][7].
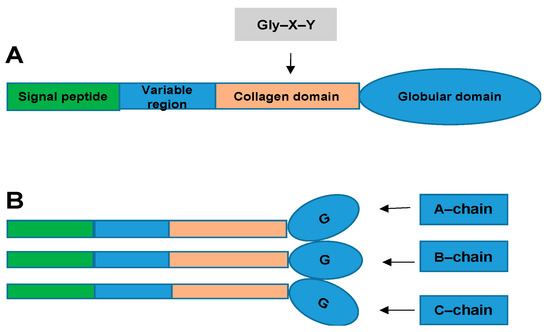
Structural organization of the C1q complement/tumor necrosis factor (TNF)-associated proteins (CTRPs). (
): Domain structure of a CTRP monomeric protein. (
): Homotrimeric CTRP protein structure. CTRP monomeric proteins form homotrimeric protein structures.
2. CTRPs and Cardiac Diseases
Growing evidence has confirmed that CTRPs exerts crucial effects on cardioprotection aspects through anti-inflammation, antiapoptosis, antifibrosis, and proangiogenesis.
2.1. CTRPs and Heart Failure
Cardiac hypertrophy is an adaptive response to maintain cardiac function, but it ultimately becomes mainly maladaptive and leads to heart failure (HF). However, accumulated data provided evidence that CTRPs are highly associated with cardiometabolics, inflammatory response, and immunoregulatory function [53,54,55][8][9][10].
CTRP3 expression is upregulated in hypertrophic and failing hearts in murine models [56][11]. CTRP3 also promotes pressure overload-induced cardiac hypertrophy via activation of the transforming growth factor β-activated kinase 1-c-Jun N-terminal kinase (TAK1-JNK) axis [56][11]. CTRP9 is upregulated in hypertrophic heart disease and induces cardiomyocyte hypertrophy and cardiac dysfunction by activating ERK5 after transverse aortic constriction (TAC) in mice [57][12]. Furthermore, this study identified GATA4 as a downstream target of CTRP9-ERK5 [57][12]. GATA4 could account for increased hypertrophy during CTRP9-ERK5 signaling, but likely not for cardiac dysfunction. Thus, additional downstream targets of the CTRP9-ERK5 probably contribute and will need to be identified. These observations advance our understanding of relationship between CTRPs and heart failure (Figure 52).

The role of CTRPs in heart failure and involved mechanisms.
2.2. CTRPs and Ischemic Cardiac Disease
The main characteristic of CTRPs are metabolic and inflammatory regulation. Accumulated data have demonstrated metabolic-related dysfunction was elicited during ischemic heart disease. Ischemic cardiac disease is a type of disease given to heart problems caused by narrowed heart arteries that lead to myocardiac ischemia (MI). A variety of links have been made between CTRPs and ischemic cardiac disease [58][13]. The involved mechanisms has been clarified in many aspects.
CTRP1 protects against myocardial ischemic injury by reducing apoptosis and inflammatory response through activation of the sphingosine-1-phosphate (S1P)/cAMP signaling pathways in cardiomyocytes, suggesting that CTRP1 plays a protective role in ischemic heart disease [59][14]. Thus, CTRP1 plays an opposite role in vascular diseases and cardiac diseases, and the underlying mechanism involved needs to be further studied. Meanwhile, research indicates CTRP3 has a protective effect on the cardiovascular system, involving multiple signaling pathways. The expression and production of CTRP3 are significantly reduced post-MI, [60][15] and replenishment of CTRP3 attenuates post-MI pathologic remodeling, including reducing heart size and cardiomyocyte apoptosis, increasing cardiomyocyte survival/regeneration, attenuating remote area interstitial fibrosis, as well as enhancing infarct border zone revascularization [60][15]. Furthermore, by promoting cardiomyocyte–endothelial cell communication involving Akt-HIF1α-VEGF signaling, CTRP3 exerts an angiogenic effect [60][15]. CTRP3 further exerts a cardiac antifibrotic effect post-MI by inhibiting myofibroblast differentiation and the subsequent extracellular matrix production. AMPK is required for the protective effect of CTRP3 against TGF-β1-induced profibrotic response. The effect of CTRP3 may be achieved by targeting the Smad3 signaling pathway [61] (Figure 3)[16]. These findings provide further understanding of the molecular mechanisms of CTRP3 in cardiac protection and provide new insights into therapeutic targets for cardiac remodeling. Perhaps preventing post-MI CTRP3 inhibition or CTRP3 supplementation can act as a promising therapeutic approach for creating stable and functional vessels post-MI, restoring cardiac function, and improving the heart failure phenotype.
CTRP6 is a cardioprotective adipokine that ameliorates ventricle remodeling post-MI, including inhibiting myofibroblast differentiation, extracellular matrix (ECM) production, and cardiac fibroblast (CF) migration [62][17]. AMPK and Akt activation contribute to the protective effect of CTRP6 against TGF-β1-induced fibrotic response by targeting the Smad-independent myocardin-related transcription factor-A (RhoA/MRTF-A) signaling pathway [62][17]. At present, there are no reports on the effects of CTRP6 on cardiomyocyte apoptosis and angiogenesis post-MI. Further study is needed on the effects of CTRP6 and its potential mechanisms. Recently, research has found that CTRP12 also ameliorates pathological remodeling of myocardium after MI by reducing myocardial inflammatory response and apoptosis in vivo [63][18]. Furthermore, CTRP12 reduces inflammatory response and apoptosis of cardiomyocytes through the PI3 kinase/Akt signaling pathway [63][18]. CTRP15, which is expressed abundantly in skeletal muscle and to a lesser extent in the lung, eye, smooth muscle, heart, and brain, exerts multiple biological effects, including promoting lipid metabolism in hepatocytes and adipocytes, suppressing autophagy in the liver, and modulating erythropoiesis [64,65,66,67,68][19][20][21][22][23]. CTRP15 is also known as myonectin [69][24]. Studies have found that CTRP15 inhibits the fibrotic response through attenuating myofibroblast differentiation and expression of profibrotic molecules on pressure overload-induced cardiac remodeling. The beneficial effects of CTRP15 on the TGF-β1-induced fibrotic response is through the IR/IRS-1/PI3K/Akt pathway. Smad3 also participates in this CTRP5′s role [70][25]. CTRP15 suppresses cardiomyocyte apoptosis and macrophage inflammatory response through the S1P-dependent activation of the cAMP/Akt pathway in the heart, thereby ameliorating acute myocardial ischemic injury [71][26]. The receptor involved in mediating these signaling pathways in cardiovascular tissues is not known and needs to be clarified in future studies (Figure 63).
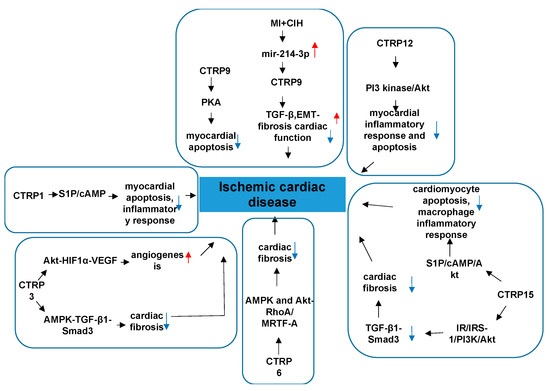
Figure 63.
The role of CTRPs in ischemic cardiac disease and involved mechanisms.
CTRP9 is the only cardiokine which is widely accepted and recognized in the CTRP family. CTRP9 is the most abundantly expressed adipokine in the heart, exceeding local APN expression more than 100-fold, with local cardiac CTRP9 levels exceeding plasma CTRP9 levels more than 2-fold [14][27]. It undergoes proteolytic cleavage to generate gCTRP9, its dominant circulatory and biologically active isoform [16][28]. Serum CTRP9 is an independent protective factor of CAD [72][29]. Serum CTRP9 decreases significantly, and the protein and mRNA expressions of CTRP9 in epicardial adipose tissue (EAT) are reduced markedly in CAD patients compared to non-CAD patients [72][29]. Although the clinical significance has been identified, basic studies clarified why CTRP9 has strong effect on the cardiac function with the following properties. Serum CTRP9 is negatively associated with traditional risk factors of cardiovascular diseases and some inflammatory factors, but it is positively associated with serum APN and high-density lipoprotein-cholesterol (HDL-C) [72][29]. These associations suggest that circulating and coronary CTRP9 play important roles in the inflammation that occurs in CAD [57][12]. CTRP9 treatment significantly decreases matrix metalloproteinase2/matrix metalloproteinase 9 (MMP2/MMP9) activity and TGF-β1 production, the two most significant mechanisms contributing to post-MI fibrosis [73][30]. CTRP9 treatment also dramatically attenuates apoptotic cell death and significantly suppresses interstitial fibrosis [73][30]. Collectively, CTRP9 prevents left ventricle (LV) remodeling by reducing apoptosis and fibrosis, and this occurs largely via a PKA-dependent pathway [73][30]. Studies in chronic intermittent hypoxia (MI+CIH) animals found that CTRP9 attenuates interstitial fibrosis, improves cardiac function, and enhances survival rate via inhibiting TGF-β/Smad and Wnt/β-catenin pathways [74][31]. MI+CIH upregulates the expression of miR-214-3p, which is the target of CTRP9 gene [74][31]. Altogether, MI+CIH suppresses cardiac CTRP9 expression by upregulating miR-214-3p, and exacerbating post-MI remodeling [74][31]. These findings provide evidence that CTRP9 and its related signaling maybe a novel therapeutic target in improving cardiac function and alleviating the heart failure (HF) phenotype in MI patients with obstructive sleep apnea (OSA). CTRP9 contributes to cardiac hypertrophy and failure during pressure overload in part through activating the ERK5-GATA4 pathway [57][12]. In the above research regarding the role of CTRP9 on remodeling post-MI, the CTRP9 was administered before or shortly after ischemia and alleviated adverse cardiac remodeling. In general, CTRP9 plays an crucial role in attenuating atrial inflammation, fibrosis, and atrial fibrillation post-MI in the following ways: it markedly downregulates inflammatory factors, interleukin-1β and interleukin-6, and upregulates interleukin-10 in 3 days post-MI to ameliorate macrophage infiltration and inflammatory responses; it reduces the expressions of collagen types I and III, a-SMA, and transforming growth factor β1 in 7 days post-MI by depressing the Toll-like receptor 4/nuclear factor-kβ and Smad2/3 signaling pathway, thus playing an antifibrotic role in the left atrium; CTRP9 ameliorates vulnerability to atrial fibrillation in post-MI rats [75][32]; it can epigenetically modulate microRNAs to adjust the genes expression. Nevertheless, it is unclear whether CTRP9 can reverse pathological immunoresponses and remodeling that have already developed. Clarification of this issue will require screening out a more delayed interval for CTRP9 administration.
2.3. CTRPs and Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DC) is a diabetes mellitus (DM)-induced pathophysiological condition that can result in HF. DC is characterized by myocardial fibrosis, dysfunctional remodeling, and associated diastolic and systolic dysfunctions, and eventually HF [76][33]. To date, there are few studies on the role of CTRPs in DC. CTRP3 in the heart protects against diabetes-related cardiac dysfunction, oxidative damage, inflammation, and cell death in vivo. Moreover, the protective effect of CTRP3 is mediated by activation of AMPKα, and CTRP3 activates AMPKα via the cAMP–EPAC–MEK–LKB1 pathways in vivo [77][34]. Its downregulation by TNF-α-initiated oxidative PPARγ suppression exacerbates cardiac injury in diabetic hearts [14][27]. These limited results may provide some theoretical basis for the role of CTRPs in DC. In addition to CTRP9, CTRP1, CTRP4, and CTRP7 are also expressed in the heart at levels significantly greater than APN. However, the impact of these CTRPs on DC are unclear. Future studies should examine the roles of these CTRPs and their underlying mechanisms in DC. (Figure 74)
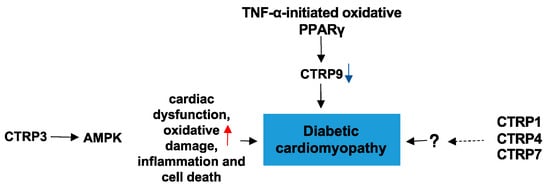
Figure 74. The role of CTRPs in diabetic cardiomyopathy and involved mechanisms.
(References would be added automatically after the entry is online)
References
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97.
- Wong, G.W.; Wang, J.; Hug, C.; Tsao, T.S.; Lodish, H.F. A family of Acrp30/adiponectin structural and functional paralogs. Proc. Natl. Acad. Sci. USA 2004, 101, 10302–10307.
- Kishore, U.; Gaboriaud, C.; Waters, P.; Shrive, A.K.; Greenhough, T.J.; Reid, K.B.; Sim, R.B.; Arlaud, G.J. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol. 2004, 25, 551–561.
- Ghai, R.; Waters, P.; Roumenina, L.T.; Gadjeva, M.; Kojouharova, M.S.; Reid, K.B.; Sim, R.B.; Kishore, U. C1q and its growing family. Immunobiology 2007, 212, 253–266.
- Schaffler, A.; Buechler, C. CTRP family: Linking immunity to metabolism. Trends Endocrinol. Metab. TEM 2012, 23, 194–204.
- Shanaki, M.; Shabani, P.; Goudarzi, A.; Omidifar, A.; Bashash, D.; Emamgholipour, S. The C1q/TNF-related proteins (CTRPs) in pathogenesis of obesity-related metabolic disorders: Focus on type 2 diabetes and cardiovascular diseases. Life Sci. 2020, 256, 117913.
- Si, Y.; Fan, W.; Sun, L. A Review of the Relationship Between CTRP Family and Coronary Artery Disease. Curr. Atheroscler. Rep. 2020, 22, 22.
- Heineke, J.; Molkentin, J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600.
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438.
- Kasher Meron, M.; Xu, S.; Glesby, M.J.; Qi, Q.; Hanna, D.B.; Anastos, K.; Kaplan, R.C.; Kizer, J.R. C1q/TNF-Related Proteins, HIV and HIV-Associated Factors, and Cardiometabolic Phenotypes in Middle-Aged Women. AIDS Res. Hum. Retroviruses 2019, 35, 1054–1064.
- Ma, Z.G.; Yuan, Y.P.; Zhang, X.; Xu, S.C.; Kong, C.Y.; Song, P.; Li, N.; Tang, Q.Z. C1q-tumour necrosis factor-related protein-3 exacerbates cardiac hypertrophy in mice. Cardiovasc. Res. 2019, 115, 1067–1077.
- Appari, M.; Breitbart, A.; Brandes, F.; Szaroszyk, M.; Froese, N.; Korf-Klingebiel, M.; Mohammadi, M.M.; Grund, A.; Scharf, G.M.; Wang, H.; et al. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ. Res. 2017, 120, 66–77.
- Zhang, Y.; Liu, C.; Liu, J.; Guo, R.; Yan, Z.; Liu, W.; Lau, W.B.; Jiao, X.; Cao, J.; Xu, K.; et al. Implications of C1q/TNF-related protein superfamily in patients with coronary artery disease. Sci. Rep. 2020, 10, 878.
- Yuasa, D.; Ohashi, K.; Shibata, R.; Mizutani, N.; Kataoka, Y.; Kambara, T.; Uemura, Y.; Matsuo, K.; Kanemura, N.; Hayakawa, S.; et al. C1q/TNF-related protein-1 functions to protect against acute ischemic injury in the heart. FASEB J. 2016, 30, 1065–1075.
- Yi, W.; Sun, Y.; Yuan, Y.; Lau, W.B.; Zheng, Q.; Wang, X.; Wang, Y.; Shang, X.; Gao, E.; Koch, W.J.; et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation 2012, 125, 3159–3169.
- Wu, D.; Lei, H.; Wang, J.Y.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Li, L.; Wu, L.L. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J. Mol. Med. 2015, 93, 1311–1325.
- Lei, H.; Wu, D.; Wang, J.Y.; Li, L.; Zhang, C.L.; Feng, H.; Fu, F.Y.; Wu, L.L. C1q/tumor necrosis factor-related protein-6 attenuates post-infarct cardiac fibrosis by targeting RhoA/MRTF-A pathway and inhibiting myofibroblast differentiation. Basic Res. Cardiol. 2015, 110, 35.
- Takikawa, T.; Ohashi, K.; Ogawa, H.; Otaka, N.; Kawanishi, H.; Fang, L.; Ozaki, Y.; Eguchi, S.; Tatsumi, M.; Takefuji, M.; et al. Adipolin/C1q/Tnf-related protein 12 prevents adverse cardiac remodeling after myocardial infarction. PLoS ONE 2020, 15, e0243483.
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J. Biol. Chem. 2012, 287, 11968–11980.
- Seldin, M.M.; Lei, X.; Tan, S.Y.; Stanson, K.P.; Wei, Z.; Wong, G.W. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J. Biol. Chem. 2013, 288, 36073–36082.
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684.
- Jiang, X.; Gao, M.; Chen, Y.; Liu, J.; Qi, S.; Ma, J.; Zhang, Z.; Xu, Y. EPO-dependent induction of erythroferrone drives hepcidin suppression and systematic iron absorption under phenylhydrazine-induced hemolytic anemia. Blood Cells Mol. Dis. 2016, 58, 45–51.
- Latour, C.; Wlodarczyk, M.F.; Jung, G.; Gineste, A.; Blanchard, N.; Ganz, T.; Roth, M.P.; Coppin, H.; Kautz, L. Erythroferrone contributes to hepcidin repression in a mouse model of malarial anemia. Haematologica 2017, 102, 60–68.
- Ouchi, N.; Walsh, K. Cardiovascular and metabolic regulation by the adiponectin/C1q/tumor necrosis factor-related protein family of proteins. Circulation 2012, 125, 3066–3068.
- Zhao, Q.; Zhang, C.L.; Xiang, R.L.; Wu, L.L.; Li, L. CTRP15 derived from cardiac myocytes attenuates TGFβ1-induced fibrotic response in cardiac fibroblasts. Cardiovasc. Drugs Ther. 2020, 34, 591–604.
- Otaka, N.; Shibata, R.; Ohashi, K.; Uemura, Y.; Kambara, T.; Enomoto, T.; Ogawa, H.; Ito, M.; Kawanishi, H.; Maruyama, S.; et al. Myonectin Is an Exercise-Induced Myokine That Protects the Heart From Ischemia-Reperfusion Injury. Circ. Res. 2018, 123, 1326–1338.
- Su, H.; Yuan, Y.; Wang, X.M.; Lau, W.B.; Wang, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.L. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res. Cardiol. 2013, 108, 315.
- Yuan, Y.; Lau, W.B.; Su, H.; Sun, Y.; Yi, W.; Du, Y.; Christopher, T.; Lopez, B.; Wang, Y.; Ma, X.L. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E891–E898.
- Wang, J.; Hang, T.; Cheng, X.M.; Li, D.M.; Zhang, Q.G.; Wang, L.J.; Peng, Y.P.; Gong, J.B. Associations of C1q/TNF-Related Protein-9 Levels in Serum and Epicardial Adipose Tissue with Coronary Atherosclerosis in Humans. BioMed. Res. Int. 2015, 2015, 971683.
- Sun, Y.; Yi, W.; Yuan, Y.; Lau, W.B.; Yi, D.; Wang, X.; Wang, Y.; Su, H.; Wang, X.; Gao, E.; et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013, 128, S113–S120.
- Du, Y.; Wang, X.; Li, L.; Hao, W.; Zhang, H.; Li, Y.; Qin, Y.; Nie, S.; Christopher, T.A.; Lopez, B.L.; et al. miRNA-Mediated Suppression of a Cardioprotective Cardiokine as a Novel Mechanism Exacerbating Post-MI Remodeling by Sleep Breathing Disorders. Circ. Res. 2020, 126, 212–228.
- Liu, M.; Li, W.; Wang, H.; Yin, L.; Ye, B.; Tang, Y.; Huang, C. CTRP9 Ameliorates Atrial Inflammation, Fibrosis, and Vulnerability to Atrial Fibrillation in Post-Myocardial Infarction Rats. J. Am. Heart Assoc. 2019, 8, e013133.
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638.
- Ma, Z.G.; Yuan, Y.P.; Xu, S.C.; Wei, W.Y.; Xu, C.R.; Zhang, X.; Wu, Q.Q.; Liao, H.H.; Ni, J.; Tang, Q.Z. CTRP3 attenuates cardiac dysfunction, inflammation, oxidative stress and cell death in diabetic cardiomyopathy in rats. Diabetologia 2017, 60, 1126–1137.