α-Amino acids find widespread applications in various areas of life and physical sciences. Their syntheses are carried out by a multitude of protocols, of which Petasis and Strecker reactions have emerged as the most straightforward and most widely used.
- amino acids
- biomolecules
- multicomponent reactions
- peptides
1. Introduction
The inadvertent discovery of the synthesis of α-amino acids by Strecker in 1850, in an attempt to prepare lactic acid from a mixture of ammonia, acetaldehyde and hydrogen cyanide [1–3][1][2][3], marked the dawn of new and stimulating advances in physical and life sciences. Indeed, following this discovery, α-amino acids, the “building blocks of life”, became readily accessible, pushing back the boundaries in peptide and protein research. This reaction, known as the “Strecker synthesis”, belongs to the general group of multicomponent reactions, and quickly became a powerful tool in the preparation of both biogenic and non-natural α-amino acids and associated substances, such as hydantoins [4,5][4][5] and polypeptides. As expected of most multicomponent reactions, the Strecker reaction allows the rapid and efficient assembly of complex α-amino acid derivatives whose preparation would otherwise be difficult, if not impossible by other methods. The simplicity of the reaction protocol as well as the possible availability of starting materials under prebiotic conditions strongly suggest that the Strecker amino acid synthesis is likely involved in the origin of life [6]. Therefore, this reaction prominently features in all discussions regarding the onset of life on earth.
α-Amino acids play a critical role in human nutrition and are extensively used as food additives thanks to their biological activity, as agrochemicals and as pharmaceuticals as well as a source for chiral building blocks in asymmetric syntheses of simple and more complex drug molecules [7,8][7][8].
In peptide and peptomimetics research, sterically hindered and α,α-disubstituted amino acids in particular have been used in numerous investigations because they induce restricted conformations when incorporated into a peptide, resulting in well-defined and pre-defined secondary structures and new properties [9]. This has guided the design of tailor-made amino acids whose integration into oligopeptides has allowed the identification of biologically active compounds such as the potent hepatitis C virus (HCV) NS3 serine protease inhibitor boceprevir [10] (see Figure 1).
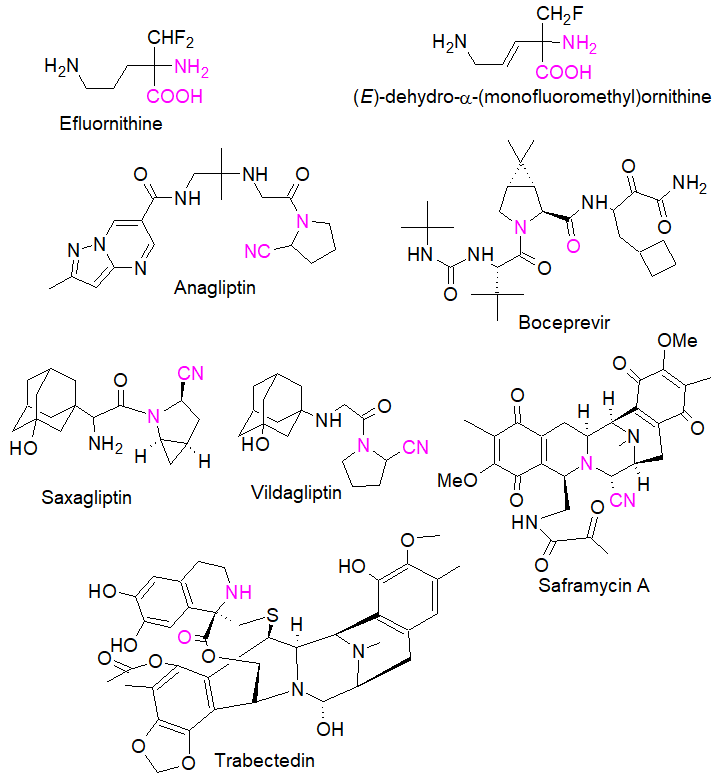
Figure 1. Examples of bioactive amino acids prepared by the Strecker synthesis. Purple colour shows the amino nitrile functions obtained as a result of a direct Strecker reaction.
Owing to all of the above reasons, the classical Strecker synthesis has gained popularity among organic chemists and remains to date the most economical and reliable method for the ready access to both natural and non-coded amino acids, and the most effective way for the preparation of large libraries of structurally diverse and distinct molecules.
The Petasis reaction, on the other hand, is relatively new, appearing in the chemistry literature in 1993, more than a century later, when Petasis and his group reported, in its original version, the reaction of a secondary amine, paraformaldehyde, and (E)-vinylboronic acid to afford the corresponding allylamines in high yields [11]. Subsequent use of glyoxylic acid as the aldehyde component resulted in the formation of the corresponding α-amino acids, paving the way for another method for the synthesis of these molecules. Like the Strecker amino acid synthesis, this reaction has all the hallmarks of all multicomponent reactions: it involves the reaction of three starting materials in a one-pot fashion to afford more complex molecules containing most, if not all the reacting components. In addition, the reaction is characterised by its simplicity, is experimentally convenient and does not require anhydrous conditions or inert atmosphere. Scheme 1 summarizes the general features of these two reactions leading to the same reaction products.
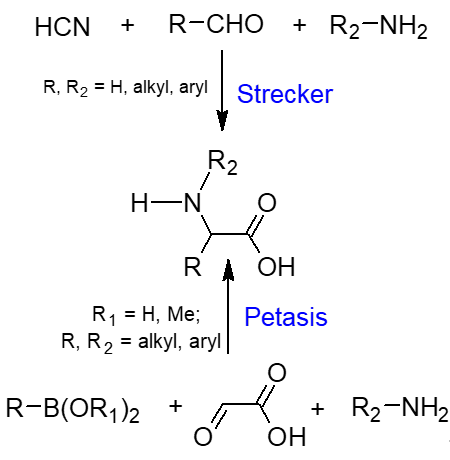
Scheme 1. General Petasis and Strecker amino acid synthesis.
There is an abundant literature regarding each of these two reactions and since they lead to the same important group of compounds, it appears relevant to compare and contrast both reactions, and outline their respective points of convergence and divergence, and delineate areas of complementarity.
2. Strecker Reaction
The classical Strecker amino acid synthesis outlined in Scheme 1 quickly developed into an industrial synthesis of both natural and non-natural racemic α-amino acids. The mechanism of the reaction is widely accepted to start by the formation of the iminium ion by the addition of ammonia (or amine) to the aldehyde or ketone; this is followed by the nucleophilic attack of the cyanide, leading to the formation of the corresponding α-aminonitrile, whose subsequent hydrolysis under basic or acidic conditions forms the amino acid (Scheme 2).
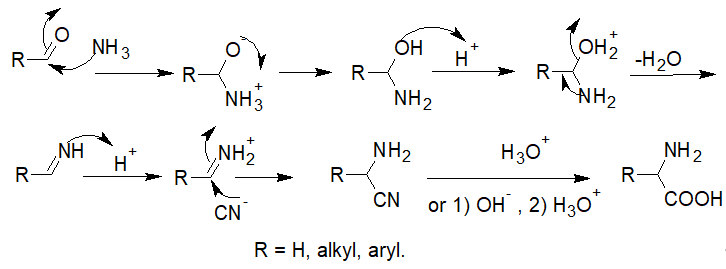
Scheme 2. General mechanism of the Strecker amino acid synthesis.
In practice and for safety purposes, solid ammonium chloride is used as the ammonia source while gaseous hydrogen cyanide is replaced by solid sodium cyanide, which is more convenient to handle.
Compared to aldehydes, the reaction with ketones is sluggish due to the additional steric hindrance brought about by the substituent replacing the hydrogen atom [12]. Products are generally obtained as a racemic mixture; nevertheless, the method remains the tool of choice in the rapid assembly of large libraries of achiral amino nitriles, whose subsequent chemical modifications and resolution produce the desired amino acids in optically pure form.
Figure 1 outlines some of the biologically relevant amino acids prepared by the direct three-component Strecker synthesis, including both conformationally rigid α- and β-diamino acids [13], reflecting the diversity and complexity of molecular entities that can be rapidly constructed by this reaction. Eflornithine and (E)-dehydro-α-monofluoromethyl)ornithine are both irreversible inhibitors of ornithine decarboxylase [14], while trabectedin is an approved drug for the treatment of cancer [15]. Saframycin A, a potent antitumor alkaloid with a pentacyclic structure, was prepared by a single step involving a Strecker reaction of three key precursors [16]; anagliptin is known to decrease the cholesterol synthesis marker lathosterol [17] whereas vildagliptin and saxagliptin, are dipeptidyl peptidase IV (DPP-IV) inhibitors [18].
The operational simplicity of the procedure has been exploited in automated processes for the synthesis of unusual amino acids such as radiolabelled [11C-carbonyl]sarcosine used in positron emission tomography (PET) imaging, starting from [11C]cyanide [19] (Scheme 3).
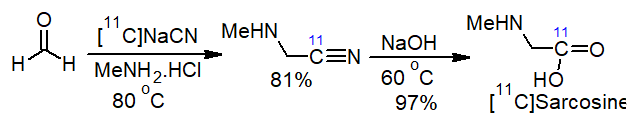
Scheme 3. Synthesis of radiolabelled [11C]sarcosine.
Beside [11C]sarcosine, the protocol was successfully applied in the production of a number of new PET radiotracers such as [11C-carbonyl]methionine, [11C-carbonyl]-N-phenylglycine and [11C-carbonyl]glycine in moderate to good radiochemical yields (79% overall yield).
Compared to earlier procedures based on the fixation of [11C]CO2 as the carbonyl source, using Grignard or organolithium reagents, the above method is clearly more straightforward and robust, whose sole limitation is the availability of the labelled cyanide.
A similar process to prepare d-[1-14C]-serine in high enantiomeric purity was employed, starting from [14C]-KCN using (R)-1-phenylethylamine as the chiral auxiliary [20].
Furthermore, membrane-active peptides can be studied under their natural conditions in lipid bilayers by solid-state NMR spectroscopy using 19F as the probe. Towards this end, Mykhailiuk et al. [21] carried out the Strecker reaction for the synthesis of the corresponding novel aliphatic 19F-substituted amino acids, which can be incorporated into bio-compatible peptides for their subsequent study by 19F-NMR spectroscopy.
The above few specific examples sufficiently demonstrate the importance of the Strecker reaction as a versatile tool in organic synthesis.
References
- Bada, J., Strecker Synthesis, in Encyclopedia of Astrobiology, M. Gargaud; et al., Editors; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011, 1603–1603, doi:10.1007/978-3-642-11274-4_1527.
- Strecker, A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Justus Liebigs Ann. Der Chem. 1850, 75, 27–45, doi:10.1002/jlac.18500750103.
- Strecker, A. Ueber einen neuen aus Aldehyd-Ammoniak und Blausäure entstehenden Körper. Justus Liebigs Ann. Der Chem. 1854, 91, 349–351, doi:10.1002/jlac.18540910309.
- Pascal, R., Bücherer–Bergs Synthesis, in Encyclopedia of Astrobiology, M. Gargaud; et al., Editors; Springer Berlin Heidelberg: Berlin, Heidelberg, 2011, 221–222, doi:10.1007/978-3-642-11274-4_210.
- Monteiro, J.L.; Pieber, B.; Corrêa, A.G.Kappe, C.O. Continuous synthesis of hydantoins: Intensifying the Bucherer–Bergs re-action. Synlett 2016, 27, 83–87, doi:10.1055/s-0035-1560317.
- Ashe, K.; Fernández-García, C.; Corpinot, M.K.; Coggins, A.J.; Bučar, D.-K.Powner, M.W. Selective prebiotic synthesis of phosphoroaminonitriles and aminothioamides in neutral water. Commun. Chem. 2019, 2, 23, doi:10.1038/s42004-019-0124-5.
- Williams, R.M.Hendrix, J.A. Asymmetric synthesis of arylglycines. Chem. Rev. 1992, 92, 889–917, doi:10.1021/cr00013a007.
- Ivanov, K.; Ivanova, S.; Georgieva, M.Atanasov, P. Production and regulatory analytical control of amino acids include in food additives. Pharmacia 2014, 61, 48–54.
- Yasufumi, O.Tetsuro, S. Asymmetric Strecker Route toward the Synthesis of Biologically Active α,α-Disubstituted α-Amino Acids. Bull. Chem. Soc. Jpn. 2003, 76, 1115–1129, doi:10.1246/bcsj.76.1115.
- Arasappan, A.; Venkatraman, S.; Padilla, A.I.; Wu, W.; Meng, T.; Jin, Y.; Wong, J.; Prongay, A.; Girijavallabhan, V.George Njoroge, F. Practical and efficient method for amino acid derivatives containing β-quaternary center: Application toward synthesis of hepatitis C virus NS3 serine protease inhibitors. Tetrahedron Lett. 2007, 48, 6343–6347, doi:10.1016/j.tetlet.2007.07.002.
- Petasis, N.A.Akritopoulou, I. The boronic acid mannich reaction: A new method for the synthesis of geometrically pure al-lylamines. Tetrahedron Lett. 1993, 34, 583–586, doi:10.1016/S0040-4039(00)61625-8.
- Hu, X.; Ma, Y.Li, Z. Eco-friendly synthesis of α-aminonitriles from ketones in PEG-400 medium using potassium Hexacy-anoferrate(II) as cyanide source. J. Organomet. Chem. 2012, 705, 70–74, doi:10.1016/j.jorganchem.2012.02.005.
- Ivon, Y.M.; Tymtsunik, A.V.; Komarov, I.V.; Shishkin, O.V.Grygorenko, O.O. Synthesis of a 2, 5-Diazabicyclo [2.2. 1] hep-tane-Derived α, β-Diamino Acid. Synth. -Stuttg. 2015, 47, 1123–1130, doi:10.1055/s-0034-1380116.
- Van Hijfte, L.; Heydt, V.Kolb, M. A versatile entry into the synthesis of α-(monofluoromethyl) amino acids: Preparation of α-(monofluoromethyl) serine and (E)-dehydro-α-(monofluoromethyl) ornithine. Tetrahedron Lett. 1993, 34, 4793–4796, doi:10.1016/S0040-4039(00)74090-1.
- Razafindrabe, C.R.; Aubry, S.; Bourdon, B.; Andriantsiferana, M.; Pellet-Rostaing, S.Lemaire, M. Synthesis of (±)-phthalascidin 650 analogue: New synthetic route to (±)-phthalascidin 622. Tetrahedron 2010, 66, 9061–9066, doi:10.1016/j.tet.2010.08.053.
- Myers, A.G.Kung, D.W. One-Step Construction of the Pentacyclic Skeleton of Saframycin A from a “Trimer” of α-Amino Aldehydes. Org. Lett. 2000, 2, 3019–3022, doi:10.1021/ol0063398.
- Aoki, K.; Ijima, T.; Kamiyama, H.; Kamiko, K.Terauchi, Y. Anagliptin decreases serum lathosterol level in patients with type 2 diabetes: A pilot study. Expert Opin. Pharmacother. 2015, 16, 1749–1754, doi:10.1517/14656566.2015.1057120.
- Augeri, D.J.; Robl, J.A.; Betebenner, D.A.; Magnin, D.R.; Khanna, A.; Robertson, J.G.; Wang, A.; Simpkins, L.M.; Taunk, P.; Huang, Q.; et al. Discovery and Preclinical Profile of Saxagliptin (BMS-477118): A Highly Potent, Long-Acting, Orally Active Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2005, 48, 5025–5037, doi:10.1021/jm050261p.
- Xing, J.; Brooks, A.F.; Fink, D.; Zhang, H.; Piert, M.R.; Scott, P.J.Shao, X. High-yielding automated convergent synthesis of no-carrier-added [11C-carbonyl]-labeled amino acids using the Strecker Reaction. Synlett: Acc. Rapid Commun. Synth. Org. Chem. 2017, 28, 371, doi:10.1055/s-0036-1588638.
- Song, F.; Salter, R.Weaner, L.E. A short synthesis of d-[1-14C]-serine of high enantiomeric purity. J. Label. Compd. Radiopharm. 2015, 58, 173–176. h, doi:10.1002/jlcr.3272.
- Bandak, D.; Babii, O.; Vasiuta, R.; Komarov, I.V.Mykhailiuk, P.K. Design and synthesis of novel 19F-amino acid: A promising 19F NMR label for peptide studies. Org. Lett. 2015, 17, 226–229, doi:10.1021/ol503300m.