Parkinson’s disease (PD) is a complex neurodegenerative disorder that is currently incurable. As a consequence of an incomplete understanding of the etiology of the disease, therapeutic strategies mainly focus on symptomatic treatment. Even though the majority of PD cases remain idiopathic (~90%), several genes have been identified to be causative for PD, facilitating the generation of animal models that are a good alternative to study disease pathways and to increase our understanding of the underlying mechanisms of PD. Drosophila melanogaster has proven to be an excellent model in these studies.
- Parkinson’s disease
- Drosophila melanogaster
- mitochondria
- endo-lysosomal pathway
- lipid home-ostasis
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease and is characterized by bradykinesia, tremor and rigidity [1][2]. These motor signs arise from a lack of dopamine due to a decline of dopaminergic neurons in the substantia nigra [3]. Furthermore, the majority of PD patients present with Lewy bodies, the pathological hallmark of PD that are aggregates of proteins–mostly consisting of alpha-synuclein-, but also containing mitochondria and lipids [4]. In addition to motor symptoms, a wide range of non-motor symptoms occur, including dementia, depression, constipation and sleep problems [5][6][7][8]. These non-motor symptoms may occur years prior to the initial diagnosis [7] and strongly impact the quality of life [9].
PD is a progressive disorder, the prevalence of which increases with age and globally affects 1% of the population above 60 years [10][11][12] imposing a heavy burden on patients, caregivers, and society. An exponential increase in PD prevalence can be expected, in part due to an increasingly aging society [13]. Hence, therapeutic strategies that halt or reverse the disease are invaluable. In the past decades, major advances have been made in our understanding of the disease pathogenesis. However, unfortunately, important gaps remain to be further elucidated and, thus, alleviating signs and symptoms via e.g., dopamine replacement therapy is currently the major therapeutic strategy [14]. To increase our understanding of the underlying pathways resulting in PD, animal models that present with the basic PD-like features, including dopaminergic neuron loss and motor signs are key. Studies in these animal models provide novel insights into disease mechanisms and identify novel therapeutic targets. Unfortunately, an animal that completely represents all signs and symptoms does not exist. Animal models created in different species often reproduce one or several PD-related signs and symptoms, while lacking other essential features. A common and remarkable feature in different genetic mouse models of PD is the lack of dopaminergic neurodegeneration [15][16][17][18], suggestive of a protective mechanism in mice that is absent in humans and pointing to the fact that each animal model has shortcomings when compared to model human diseases.
Nonetheless, Drosophila melanogaster is an excellent model to study the molecular mechanisms in PD. The fruit fly is a relatively simple animal; however, it does contain a complex neuronal circuitry including clusters of dopaminergic neurons [19][20]. D. melanogaster undergoes different stages (egg, larva, pupa, fly) in a short lifecycle of around 10 days (Table 1). Furthermore, many experimental tools exist to genetically manipulate D. melanogaster allowing a relatively rapid creation of genetically modified flies that can serve as a disease model. In this review, we focus on the advantages of using D. melanogaster as animal model to study disease pathways and the key findings in the molecular mechanisms in PD that arose from studies on the fruit fly.
Table 1. Limitations and advantages of different aspects of using D. melanogaster as animal model to study human diseases.
|
Feature |
Limitation |
Advantage |
|
Life cycle of ~10 days |
Too short to study late-life stage signs |
A lot of flies in a short amount of time |
|
Behavior |
Not all aspects can be analyzed |
Locomotion, sleep, circadian rhythm, can be analyzed |
|
Brain |
Neuronal circuitry is not evolutionarily conserved |
Complex neuronal circuitry (including dopaminergic neurons) |
|
UAS-gal4 system |
Off-target effects Overexpression not controlled: too much protein, and thus less physiological condition |
Overexpression of human disease genes Knockdown of genes to mimic a loss of function |
|
Genome |
Only 4 chromosomes versus 23 in human |
75% of the disease-causing genes have a fly ortholog |
2. Drosophila melanogaster as Animal Model to Study Human Diseases
Fruit flies are small animals, which facilitates live imaging enabling in vivo recording of biological processes that can further clarify disease-relevant mechanisms, including analyzing dopaminergic neurons that are mainly affected in PD (Table 1). The genome of flies consists of only four chromosomes, which is a markedly lower number than the 23 chromosomes in the human genome. Nonetheless, more than 75% of the human disease-causing genes have an ortholog in flies (Table 1) [21]. Using established methodology to manipulate genes, including the Crispr cas9 system that allows the introduction of specific point mutations [22][23] or the yeast UAS-gal4 system enabling overexpression or knockdown of proteins ubiquitously or in relevant tissue (Table 1) [24][25][26], the proper genetic allele can be created that mimics the genetic disease context to study essential biological processes. For example, overexpression of a specific protein may be more relevant for gain-of-function mutations than knocking down that specific protein. In addition, several collections of mutant flies exist or can be created via toxin-induced mutagenesis. These are especially useful to perform modifier screens, which is one of the benefits of using D. melanogaster in the study of diseases. These modifier screens enable the identification of genes that interact with disease-related mechanisms contributing to the elucidation of novel pathways involved in or providing promising therapeutic targets. Such modifier screens have proven their value in PD research with the identification of vitamin K2, aconitase, sterol regulatory element-binding protein 1 (SREBP1) (see below) [27][28][29]. Furthermore, screens in which specific phenotypes are analyzed that mimic phenotypes of a specific disease model can lead to the identification of new genes that are involved in the disease pathogenesis (Figure 1).
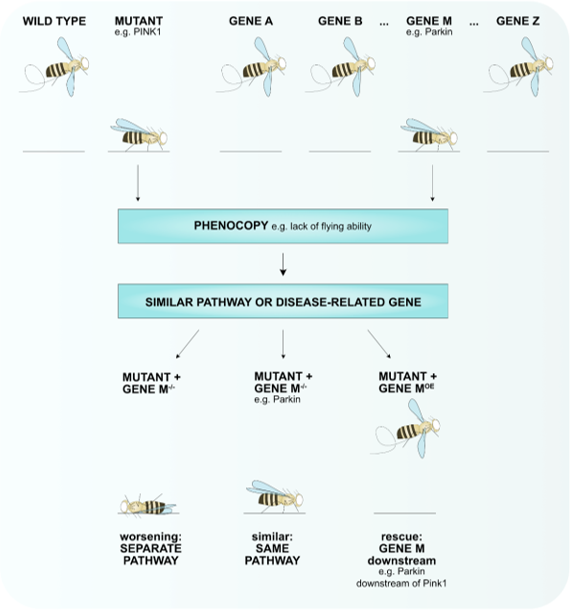
Figure 1. A phenotypic screen can identify novel genes involved in the disease pathway. A collection of mutant alleles (gene A–gene Z) can be used to identify genes that when mutated result in the same phenotype (phenocopy) e.g., lack of flying ability for pink1-mutant flies that are mimicked by parkin-mutant flies. To test if these genes function in the same pathway, the phenotypes of double-mutant flies can be tested. Loss of gene M worsens the phenotype of the disease mutant, thus, separate pathways play a role that converge into one. Loss of gene M does not enhance or improve the phenotype of the mutant meaning that these genes function in the same pathway (e.g., Pink1-Parkin). Overexpression (OE) of gene M rescues the mutant phenotype hints towards gene M functioning downstream of the mutant (e.g., Parkin functions downstream of Pink1).
The creation of new fly models often emerges from the identification of a disease-causing gene. For example, loss-of-function mutations in Thousand and one amino acid kinase 1 (TOAK1) have been identified to be causative for neurodevelopmental delay and intellectual disability [30]. Knockdown of the Drosophila ortholog Tao shows a delayed development and lethality at the final larval stage accompanying defective brain and neuronal morphology [30] mimicking the disease phenotype. Furthermore, knockdown of Tao presents with altered mitochondrial distribution in axons of motor neurons [30], suggesting mitochondria play a role in the disease pathogenesis, which was confirmed in patient-derived fibroblasts that display defective mitochondrial morphology [30]. Thus, this fly model for neurodevelopmental delay can be used to further investigate the relationship between abnormal mitochondria and brain morphology that results in neurodevelopmental delay. Notably, D. melanogaster can also be used to model diseases that do not occur naturally in flies. Lethal tumor growth and metastasis have not been observed in wild type flies [21]. Nonetheless, genes that play a role in tumor formation in humans, such as those affecting the cell-cycle control have been identified in flies and are being used to study processes of tumor growth [21]. These examples show that D. melanogaster can be employed to study a plethora of human diseases even if the exact disease process does not occur in this animal.
References
- Fahn, S.; Jankovic, J. Principles and Practice of Movement Disorders; Churchill Livingstone Elsevier: Philadelphia, PA, USA, 2007.
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741, doi:10.1002/mds.27352.
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 715–724.
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109, doi:10.1038/s41593-019-0423-2.
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245.
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435.
- Lee, H.; Koh, S. Many Faces of Parkinson’s Disease: Non-Motor Symptoms of Parkinson’s Disease. Mov. Disord. 2015, 8, 92–97.
- Kasten, M.; Marras, C.; Klein, C. Nonmotor Signs in Genetic Forms of Parkinson’s Disease. In International Review of Neurobi-ology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 129–178, ISBN 9780128137086.
- Hermanowicz, N.; Jones, S.A.; Hauser, R.A. Impact of non-motor symptoms in Parkinson’s disease: A PMDAlliance survey. Neuropsychiatr. Dis. Treat. 2019, 15, 2205–2212, doi:10.2147/NDT.S213917.
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10.
- GBD Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480.
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535.
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, in press.
- You, H.; Mariani, L.; Mangone, G.; Le Febvre de Nailly, D.; Charbonnier-Beaupel, F.; Corvol, J. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. Cell Tissue Res. 2018, 373, 111–135.
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Müller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterio-ration and in human alpha-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439, doi:10.1172/JCI15777.
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.Y. Neuronal [alpha]-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human [alpha]-Synuclein. Neuron 2002, 34, 521–533.
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS ONE 2009, 4, e5777–e5777, doi:10.1371/journal.pone.0005777.
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Cepeda, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; et al. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635.
- Yamamoto, S.; Seto, E. Dopamine dynamics and signaling in Drosophila: An overview of genes, drugs and behavioral para-digms. Exp. Anim. 2014, 63, 107–119.
- Nagoshi, E. Drosophila Models of Sporadic Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 3343.
- Bier, E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 2005, 6, 9–23.
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193.
- Gratz, S.; Cummings, A.; Nguyen, J.; Hamm, D.; Donohue, L.; Harrison, M.; Wildonger, J.; O’Connor-Giles, K. Genome en-gineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. Genetics 2013, 194, 1029–1035.
- Webster, N.; Jin, J.; Green, S.; Hollis, M.; Chambon, P. The yeast UASG is a transcriptional enhancer in human HeLa cells in the presence of the GAL4 trans-activator. Cell 1988, 52, 169–178.
- Kakidani, H.; Ptashne, M. GAL4 activates gene expression in mammalian cells. Cell 1988, 52, 161–167.
- Duffy JB GAL4 system in Drosophila: A fly geneticist’s Swiss army knife. Genes Brain Behav. 2002, 34, 1–15.
- Vos, M.; Esposito, G.; Edirisinghe, J.N.; Vilain, S.; Haddad, D.M.; Slabbaert, J.R.; Van Meensel, S.; Schaap, O.; De Strooper, B.; Meganathan, R.; et al. Vitamin K2 is a mitochondrial electron carrier that rescues pink1 deficiency. Science 2012, 336, 1306–1310.
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; De Sousa Valadas, J.; Van Meensel, S.; Schaap, O.; Verstreken, P. Van Aconitase Causes Iron Toxicity in Drosophila pink1 Mutants. PLoS Genet. 2013, 9, e1003478.
- Ivatt, R.; Sanchez-Martinez, A.; Godena, V.; Brown, S.; Ziviani, E.; Whitworth, A. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499.
- Dulovic-Mahlow, M.; Trinh, J.; Kandaswamy, K.K.; Braathen, G.J.; Di Donato, N.; Rahikkala, E.; Beblo, S.; Werber, M.; Krajka, V.; Busk, Ø.L.; et al. De Novo Variants in TAOK1 Cause Neurodevelopmental Disorders. Am. J. Hum. Genet. 2019, 105, 213–220, doi:10.1016/j.ajhg.2019.05.005.