T-lymphokine-activated killer cell-originated protein kinase (TOPK), also known as PDZ-binding kinase (PBK), was a member of the MEK3/6-related MAPKK family. As a mitotic serine/threonine protein kinase, accumulating evidence supported its role in mitosis and cell-cycle progression of mitotically active cells, especially proliferative malignant cells. PBK/TOPK was confirmed to be associated with the development, progression, and metastasis of malignancies, which made it a potential therapeutic target in cancer therapy. Further, it was also demonstrated to play crucial roles in ischemic injury and involve in protection against ischemia. This protective effect of PBK/TOPK in the context of ischemia challeged the development of PBK/TOPK inhibitors in anti-tumor therapy, and more research was required to further explore its role and underlying mechanisms to translate its application to clinical studies.
- PBK/TOPK
- mitosis
- malignancy
- ischemia
- inhibitor
1. Introduction
T-lymphokine-activated killer-cell-originated protein kinase (TOPK), also known as PDZ-binding kinase (PBK), is a novel mitotic serine/threonine protein kinase [1,2][1][2]. PBK/TOPK is overexpressed in various actively proliferative cells, including malignant tumor cells, as well as normal cells, such as sperm cells. The transcription, activation, and degradation of PBK/TOPK is regulated by mitosis progression, which is mediated by a series of proteins. As a mitotic kinase, PBK/TOPK is important for the proliferation, progression, and metastasis of many cancers, including leukemia and myeloma, among others [3,4][3][4]. Upregulation of PBK/TOPK has also been proven to be associated with cancer diagnosis and prognosis, and, thus, it may be a potential therapeutic target in various malignant tumors. Thus, various potent PBK/TOPK inhibitors have been developed. The function of PBK/TOPK as an emerging target for cancer-specific therapeutics has been reviewed previously by Herbert et al. [5]. Furthermore, ours and other research teams also demonstrated that PBK/TOPK plays crucial roles in ischemic injury and is involved in protection against ischemia and ischemic postconditioning [6,7,8][6][7][8]. This protective effect of PBK/TOPK in the context of ischemia challenged the development of PBK/TOPK inhibitors in antitumor therapy, and more research is required to further explore its role and underlying mechanisms, to translate its applicability to clinical studies.
2. Identification of PBK/TOPK
The human homologue of the Drosophila tumor suppressor Discs-large (hDlg) is a membrane-associated guanylate kinase homologue (MAGUK) [9]. Several proteins that bind to hDlg have been demonstrated to be involved in cell growth control, including in the pathogenesis of adenomatous polyposis coli [10,11,12,13][10][11][12][13]. In 2000, Gaudet et al. sought to elucidate the signal transduction pathway through which hDlg regulates cellular proliferation, and they first identified a 322 amino acid serine/threonine kinase, using a two-hybrid screen and named it PDZ-binding kinase (PBK) [2]. At the same time, Abe et al. cloned a novel protein kinase from a lymphokine-activated killer T (T-LAK) cell subtraction cDNA fragment library and named it T-LAK cell-originated protein kinase (TOPK) [1]. This gene was later confirmed to be similar to that found by Gaudet et al., and this novel gene, or its protein product, was called PBK/TOPK.
3. General Features of PBK/TOPK
Unlike hDlg, which is ubiquitously expressed, the expression and distribution of PBK/TOPK varies. It is abundant in highly proliferative tissues, such as the placenta, testis, T-LAK cells, activated lymphoid cells and lymphoid tumors, but it is expressed at a very low level or is even absent in non-proliferative normal tissues, such as normal adult brain tissue (with the exception of the subependymal zone and early postnatal cerebellar external layer, which is enriched with rapidly proliferating progenitor cells) [14,15][14][15]. Regarding its intracellular distribution, PBK/TOPK is expressed both in the cytoplasm and the nucleus; however, it may be expressed exclusively in the nucleus of tumor cells or in mitosis, particularly around chromosomal surfaces during prophase and metaphase [16,17][16][17]. Moreover, both PBK/TOPK protein expression and its activation are subject to cell-cycle regulation. Using the thymidine double-block method, PBK/TOPK protein expression was demonstrated to increase and peak at 8 h after being released from the cell-cycle block during G2 to M phase in HeLa cells, and this was accompanied by the upregulation of its phosphorylation activity [18].
As a member of the MEK3/6-related MAPKK family, PBK/TOPK shares the characteristic serine/threonine protein kinase subdomains and a C-terminal PDZ-binding T/SXV motif, which enables it to bind specifically to the PDZ2 domain of hDlg or other PDZ-containing proteins. Activated PBK/TOPK is also able to phosphorylate P38 MAPK and histone H3, among other proteins [2,19,20][2][19][20]. However, the phosphotransferase activity of PBK/TOPK appears to be regulated in a cell-cycle-dependent manner; that is, it is activated following its phosphorylation by cyclin-dependent kinase 1 (CDK1)/cyclin B1 exclusively at mitosis (Figure 1). Notably, this process depends on the binding of PBK/TOPK to the CDK1/cyclin B1 complex at the mitotic spindle, which in turn induces its phosphorylation and thereby increases its binding ability to the CDK1/cyclin B1 complex through phosphorylation and inactivation of protein phosphatase 1 alpha (PP1α) [2,21,22][2][21][22]. These results all suggest that PBK/TOPK may be involved in the regulation of cellular proliferation and cell-cycle progression.
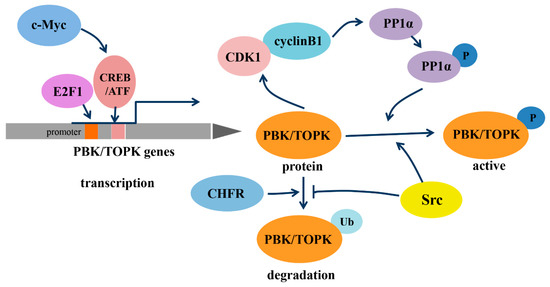
Regulation of transcription, phosphorylation and degradation of PDZ-binding kinase/T-lymphokine-activated killer-cell-originated protein kinase (PBK/TOPK). Abbreviations: c-Myc, the human cellular homologue of the v-myc oncogene of avian myelocytomatosis retrovirus MC29; E2F1, transcription factor E2F contained eight E2F genes, including E2F1-8; CREB/ATF, cyclic AMP-responsive element binding protein/activating transcription factor; CDK1, cyclin-dependent kinase 1; PP1α, protein phosphatase 1 alpha; CHFR, checkpoint protein with FHA and RING domains; Src, a non-receptor tyrosine kinase.
In addition, the transcription of PBK/TOPK and its degradation varies with cell-cycle progression (Figure 1). The binding of the cell-cycle-specific transcription factors E2F and cyclic AMP-responsive element-binding protein/activating transcription factor (CREB/ATF) to the −146 and −312 bp binding sites within the PBK/TOPK promoter directly leads to its transcriptional upregulation [17,23][17][23]. C-Myc is functionally linked with E2F1 in controlling cell-cycle progression, and it activates PBK/TOPK transcription by indirectly enhancing E2F1 activity and cooperatively binding with E2F1 [24]. Checkpoint protein with FHA and RING domains (CHFR) is an E3 ubiquitin ligase that was demonstrated to ubiquitinate and mediate PBK/TOPK degradation, which is essential for its mitotic checkpoint function [25]. Src, a transforming protein with tyrosine kinase activity, was also identified as an upstream regulator of PBK/TOPK. It can directly bind and phosphorylate PBK/TOPK at Y74 and Y272, thereby enhancing its activity and enhancing PBK/TOPK stability, which allows it to avoid degradation after ubiquitination [26].
4. PBK/TOPK Function in Mitotic Progression and Tumor Cellular Proliferation
Mitotic progression is regulated by the co-ordination of several proteins and is crucial for the maintenance of genomic stability. PBK/TOPK is known to play pivotal roles in cellular mitosis; its main mitotic substrates and detailed functions in mitosis have been studied in depth (Figure 2). PBK/TOPK is of crucial importance in CHFR-mediated G2/M progression via phosphorylation and inactivation of PTEN, which results in activation of Akt [25]. Activated TOPK was also confirmed as the principal C2H2 zinc finger protein (ZFP) linker kinase that phosphorylates C2H2 ZFPs linkers within minutes in the prophase stage. This is a unique mechanism for reducing the DNA-binding activity of C2H2 ZFPs, which mediates the dissociation of hundreds of proteins from condensed chromatin during transition from prophase to telophase. PBK/TOPK was the first kinase to be identified as a master regulator of an entire family of transcription factors, based on their conserved motif [27]. At the late stage of mitosis, upregulated PBK/TOPK was shown to mediate the phosphorylation of LGN/GPSM2 (Leu-Gly-Asn repeat-enriched protein/G-protein signaling modulator 2) at Thr450, which might play a critical role in the segregation of sister chromatids and the formation of F-actin polymerization at the contractile ring [28]. In addition, activated PBK/TOPK was demonstrated to enhance the phosphorylation of the microtubule-binding protein PRC1 and ATPase protein p97 with p47 as an adaptor protein, which is indispensable for mitotic spindle formation and cytokinesis during mitosis [29].
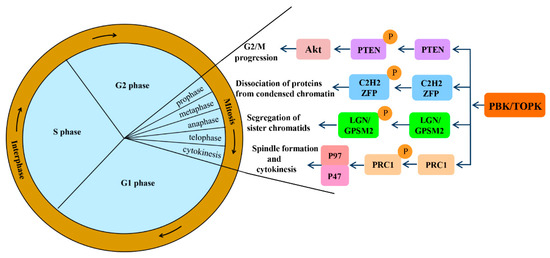
Signaling pathways involved in mitotic progression functions of PBK/TOPK. Abbreviations: PTEN, phosphatase and tensin homolog; Akt, also known as protein kinase B; ZFP, zinc finger proteins; LGN/GPSM2, Leu-Gly-Asn repeat-enriched protein/G-protein signaling modulator 2; PRC1, microtubule binding protein.
PBK/TOPK overexpression in oncogenic pathways has been implicated in the proliferation, metastasis, and anti-apoptosis of tumors (Figure 3). Its overexpression allows the tumor cells to bypass the natural surveillance mechanism associated with the G2/M checkpoint and leads to aberrant entry into the mitotic phase by phosphorylating histone H3 at Ser10, as well as down-modulating the tumor suppressor p53, upregulating the cyclin-dependent kinase inhibitor p21, and thereby contributing to tumorigenesis [30,31,32][30][31][32]. Conversely, suppression of PBK/TOPK impaired its tumorigenic properties by reducing the phosphorylation and activation of mitogen-activated protein (MAP) kinase, including extracellular signal-regulated kinase (ERK) 2 and P38, inhibiting the expression of mutant p53, and inhibiting Akt activation [33,34,35,36,37,38][33][34][35][36][37][38]. Notably, phosphorylated ERK2 could in turn phosphorylate PBK/TOPK and increase its kinase activity, resulting in a positive feedback loop between PBK/TOPK and ERK2. PBK/TOPK also possesses the capacity to activate the interaction of transcriptional factor β-catenin, with its transcriptional coactivators T-cell factor/lymphoid enhancer-binding factor (TCF/LEF), which subsequently upregulates the transcription of matrix metalloproteinase MMP-2 and MMP-9, thereby facilitating the invasiveness and metastasis of tumor cells [16].

Signaling pathways involved in tumorigenic functions of PBK/TOPK. Abbreviations: H3, histone H3; ERK2, extracellular signal-regulated kinase 2; TCF/LEF, T-cell factor/lymphoid enhancer–binding factor; MMP-2/9, matrix metalloproteinase-2/9; JNK1, c-Jun-NH2-Kinase 1; Prx1, peroxiredoxin1; IκBα, inhibitor-κBα; NF-κB, nuclear factor kappa B.
Aside from promoting proliferation and metastasis, activated PBK/TOPK could also alleviate As3+ treatment–induced apoptosis by binding with and phosphorylating histone H2AX at Ser139 in the nucleus. This may be responsible for the resistance of some melanoma cells to As3+ treatment [39]. Moreover, PBK/TOPK could inhibit UVB-induced apoptosis as well, by phosphorylating c-Jun-NH2-Kinase 1 (JNK1) at Thr183/Tyr185, activating it, and increasing the peroxidase activity of peroxiredoxin1 (Prx1) and decreasing the intracellular accumulation of H2O2 via phosphorylation Prx1 at Ser32 [40,41][40][41]. Moreover, TOPK directly phosphorylates inhibitor-κBα (IκBα) at Ser 32, which induces P65 nuclear translocation, nuclear factor-kappa B (NF-κB) activation, and subsequently leads to responsive transcription of anti-apoptotic genes [42,43,44][42][43][44]. These findings enrich our knowledge of the regulatory functions and underlying mechanisms of PBK/TOPK in mitotic progression of actively proliferating cells, especially tumors, and have led to a better understanding of its role in tumorigenesis. These studies all suggest that PBK/TOPK might serve as a diagnostic/prognostic indicator and therapeutic target in tumors.
References
- Abe, Y.; Matsumoto, S.; Kito, K.; Ueda, N. Cloning and expression of a novel MAPKK-like protein kinase, lymphokine-activated killer T-cell-originated protein kinase, specifically expressed in the testis and activated lymphoid cells. J. Biol. Chem. 2000, 275, 21525–21531.
- Gaudet, S.; Branton, D.; Lue, R.A. Characterization of PDZ-binding kinase, a mitotic kinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5167–5172.
- Ikeda, Y.; Park, J.H.; Miyamoto, T.; Takamatsu, N.; Kato, T.; Iwasa, A.; Okabe, S.; Imai, Y.; Fujiwara, K.; Nakamura, Y.; et al. T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 6110–6117.
- Ishikawa, C.; Senba, M.; Mori, N. Mitotic kinase PBK/TOPK as a therapeutic target for adult Tcell leukemia/lymphoma. Int. J. Oncol. 2018, 53, 801–814.
- Herbert, K.J.; Thomas, M.A.; Remko, P.; Giacomo, P.; Geoff, S.H. T-LAK cell-originated protein kinase (TOPK): An emerging target for cancer-specific therapeutics. Cell Death Dis. 2018, 9, 1089.
- Zhao, H.; Wang, R.; Tao, Z.; Gao, L.; Yan, F.; Gao, Z.; Liu, X.; Ji, X.; Luo, Y. Ischemic postconditioning relieves cerebral ischemia and reperfusion injury through activating T-LAK cell-originated protein kinase/protein kinase B pathway in rats. Stroke 2014, 45, 2417–2424.
- Sun, G.; Ye, N.; Dai, D.; Chen, Y.; Li, C.; Sun, Y. The Protective Role of the TOPK/PBK Pathway in Myocardial Ischemia/Reperfusion and H(2)O(2)-Induced Injury in H9C2 Cardiomyocytes. Int. J. Mol. Sci. 2016, 17, 267.
- Gao, S.; Zhu, Y.; Li, H.; Xia, Z.; Wu, Q.; Yao, S.; Wang, T.; Yuan, S. Remote ischemic postconditioning protects against renal ischemia/reperfusion injury by activation of T-LAK-cell-originated protein kinase (TOPK)/PTEN/Akt signaling pathway mediated anti-oxidation and anti-inflammation. Int. Immunopharmacol. 2016, 38, 395–401.
- Lue, R.A.; Marfatia, S.M.; Branton, D.; Chishti, A.H. Cloning and characterization of hdlg: The human homologue of the Drosophila discs large tumor suppressor binds to protein 4.1. Proc. Natl. Acad. Sci. USA 1994, 91, 9818–9822.
- Matsumine, A.; Ogai, A.; Senda, T.; Okumura, N.; Satoh, K.; Baeg, G.H.; Kawahara, T.; Kobayashi, S.; Okada, M.; Toyoshima, K.; et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science 1996, 272, 1020–1023.
- Li, X.; Lin, S.; Chen, X.; Huang, W.; Li, Q.; Zhang, H.; Chen, X.; Yang, S.; Jin, K.; Shao, B. The Prognostic Value of Serum Cytokines in Patients with Acute Ischemic Stroke. Aging Dis. 2019, 10, 544–556.
- Liu, D.; Xu, L.Q.; Zhang, X.Y.; Shi, C.H.; Qiao, S.B.; Ma, Z.Q.; Yuan, J.S. Snapshot: Implications for mTOR in Aging-related Ischemia/Reperfusion Injury. Aging Dis. 2019, 10, 116–133.
- Shen, F.X.; Jiang, L.D.; Han, F.; Degos, V.; Chen, S.D.; Su, H. Increased Inflammatory Response in Old Mice is Associated with More Severe Neuronal Injury at the Acute Stage of Ischemic Stroke. Aging Dis. 2019, 10, 12–22.
- Fujibuchi, T.; Abe, Y.; Takeuchi, T.; Ueda, N.; Shigemoto, K.; Yamamoto, H.; Kito, K. Expression and phosphorylation of TOPK during spermatogenesis. Dev. Growth Differ. 2005, 47, 637–644.
- Dougherty, J.D.; Garcia, A.D.; Nakano, I.; Livingstone, M.; Norris, B.; Polakiewicz, R.; Wexler, E.M.; Sofroniew, M.V.; Kornblum, H.I.; Geschwind, D.H. PBK/TOPK, a proliferating neural progenitor-specific mitogen-activated protein kinase kinase. J. Neurosci. 2005, 25, 10773–10785.
- Brown-Clay, J.D.; Shenoy, D.N.; Timofeeva, O.; Kallakury, B.V.; Nandi, A.K.; Banerjee, P.P. PBK/TOPK enhances aggressive phenotype in prostate cancer via beta-catenin-TCF/LEF-mediated matrix metalloproteinases production and invasion. Oncotarget 2015, 6, 15594–15609.
- Chen, J.H.; Liang, Y.X.; He, H.C.; Chen, J.Y.; Lu, J.M.; Chen, G.; Lin, Z.Y.; Fu, X.; Ling, X.H.; Han, Z.D.; et al. Overexpression of PDZ-binding kinase confers malignant phenotype in prostate cancer via the regulation of E2F1. Int. J. Biol. Macromol. 2015, 81, 615–623.
- Matsumoto, S.; Abe, Y.; Fujibuchi, T.; Takeuchi, T.; Kito, K.; Ueda, N.; Shigemoto, K.; Gyo, K. Characterization of a MAPKK-like protein kinase TOPK. Biochem. Biophys. Res. Commun. 2004, 325, 997–1004.
- Yuryev, A.; Wennogle, L.P. Novel raf kinase protein-protein interactions found by an exhaustive yeast two-hybrid analysis. Genomics 2003, 81, 112–125.
- Li, S.; Zhu, F.; Zykova, T.; Kim, M.O.; Cho, Y.Y.; Bode, A.M.; Peng, C.; Ma, W.; Carper, A.; Langfald, A.; et al. T-LAK cell-originated protein kinase (TOPK) phosphorylation of MKP1 protein prevents solar ultraviolet light-induced inflammation through inhibition of the p38 protein signaling pathway. J. Biol. Chem. 2011, 286, 29601–29609.
- Park, J.H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Critical roles of T-LAK cell-originated protein kinase in cytokinesis. Cancer Sci. 2010, 101, 403–411.
- Cote, S.; Simard, C.; Lemieux, R. Regulation of growth-related genes by interleukin-6 in murine myeloma cells. Cytokine 2002, 20, 113–120.
- Nandi, A.K.; Rapoport, A.P. Expression of PDZ-binding kinase (PBK) is regulated by cell cycle-specific transcription factors E2F and CREB/ATF. Leuk. Res. 2006, 30, 437–447.
- Hu, F.; Gartenhaus, R.B.; Zhao, X.F.; Fang, H.B.; Minkove, S.; Poss, D.E.; Rapoport, A.P. c-Myc and E2F1 drive PBK/TOPK expression in high-grade malignant lymphomas. Leuk. Res. 2013, 37, 447–454.
- Shinde, S.R.; Gangula, N.R.; Kavela, S.; Pandey, V.; Maddika, S. TOPK and PTEN participate in CHFR mediated mitotic checkpoint. Cell Signal. 2013, 25, 2511–2517.
- Xiao, J.; Duan, Q.; Wang, Z.; Yan, W.; Sun, H.; Xue, P.; Fan, X.; Zeng, X.; Chen, J.; Shao, C.; et al. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon. Oncotarget 2016, 7, 24483–24494.
- Rizkallah, R.; Batsomboon, P.; Dudley, G.B.; Hurt, M.M. Identification of the oncogenic kinase TOPK/PBK as a master mitotic regulator of C2H2 zinc finger proteins. Oncotarget 2015, 6, 1446–1461.
- Fukukawa, C.; Ueda, K.; Nishidate, T.; Katagiri, T.; Nakamura, Y. Critical roles of LGN/GPSM2 phosphorylation by PBK/TOPK in cell division of breast cancer cells. Genes Chromosomes Cancer 2010, 49, 861–872.
- Abe, Y.; Takeuchi, T.; Kagawa-Miki, L.; Ueda, N.; Shigemoto, K.; Yasukawa, M.; Kito, K. A mitotic kinase TOPK enhances Cdk1/cyclin B1-dependent phosphorylation of PRC1 and promotes cytokinesis. J. Mol. Biol. 2007, 370, 231–245.
- Nandi, A.K.; Ford, T.; Fleksher, D.; Neuman, B.; Rapoport, A.P. Attenuation of DNA damage checkpoint by PBK, a novel mitotic kinase, involves protein-protein interaction with tumor suppressor p53. Biochem. Biophys. Res. Commun. 2007, 358, 181–188.
- Hu, F.; Gartenhaus, R.B.; Eichberg, D.; Liu, Z.; Fang, H.B.; Rapoport, A.P. PBK/TOPK interacts with the DBD domain of tumor suppressor p53 and modulates expression of transcriptional targets including p21. Oncogene 2010, 29, 5464–5474.
- Park, J.H.; Lin, M.L.; Nishidate, T.; Nakamura, Y.; Katagiri, T. PDZ-binding kinase/T-LAK cell-originated protein kinase, a putative cancer/testis antigen with an oncogenic activity in breast cancer. Cancer Res. 2006, 66, 9186–9195.
- Ayllon, V.; O’Connor, R. PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response. Oncogene 2007, 26, 3451–3461.
- Chen, F.; Li, R.; Wang, C.; Cao, L.; Wang, Y.; Yu, L. T-LAK cell-originated protein kinase is essential for the proliferation of hepatocellular carcinoma SMMC-7721 cells. Cell Biochem. Funct. 2013, 31, 736–742.
- Yu, Y.; Bai, F.; Liu, Y.; Yang, Y.; Yuan, Q.; Zou, D.; Qu, S.; Tian, G.; Song, L.; Zhang, T.; et al. Fibroblast growth factor (FGF21) protects mouse liver against D-galactose-induced oxidative stress and apoptosis via activating Nrf2 and PI3K/Akt pathways. Mol. Cell. Biochem. 2015, 403, 287–299.
- Lei, B.; Liu, S.; Qi, W.; Zhao, Y.; Li, Y.; Lin, N.; Xu, X.; Zhi, C.; Mei, J.; Yan, Z.; et al. PBK/TOPK expression in non-small-cell lung cancer: Its correlation and prognostic significance with Ki67 and p53 expression. Histopathology 2013, 63, 696–703.
- Lei, B.; Qi, W.; Zhao, Y.; Li, Y.; Liu, S.; Xu, X.; Zhi, C.; Wan, L.; Shen, H. PBK/TOPK expression correlates with mutant p53 and affects patients’ prognosis and cell proliferation and viability in lung adenocarcinoma. Hum. Pathol. 2015, 46, 217–224.
- Zhu, F.; Zykova, T.A.; Kang, B.S.; Wang, Z.; Ebeling, M.C.; Abe, Y.; Ma, W.Y.; Bode, A.M.; Dong, Z. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology 2007, 133, 219–231.
- Zykova, T.A.; Zhu, F.; Lu, C.; Higgins, L.; Tatsumi, Y.; Abe, Y.; Bode, A.M.; Dong, Z. Lymphokine-activated killer T-cell-originated protein kinase phosphorylation of histone H2AX prevents arsenite-induced apoptosis in RPMI7951 melanoma cells. Clin. Cancer Res. 2006, 12, 6884–6893.
- Oh, S.M.; Zhu, F.; Cho, Y.Y.; Lee, K.W.; Kang, B.S.; Kim, H.G.; Zykova, T.; Bode, A.M.; Dong, Z. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer Res. 2007, 67, 5186–5194.
- Zykova, T.A.; Zhu, F.; Vakorina, T.I.; Zhang, J.; Higgins, L.A.; Urusova, D.V.; Bode, A.M.; Dong, Z. T-LAK cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of Prx1 peroxidase activity. J. Biol. Chem. 2010, 285, 29138–29146.
- Kwon, H.R.; Lee, K.W.; Dong, Z.; Lee, K.B.; Oh, S.M. Requirement of T-lymphokine-activated killer cell-originated protein kinase for TRAIL resistance of human HeLa cervical cancer cells. Biochem. Biophys. Res. Commun. 2010, 391, 830–834.
- Park, J.H.; Yoon, D.S.; Choi, H.J.; Hahm, D.H.; Oh, S.M. Phosphorylation of IkappaBalpha at serine 32 by T-lymphokine-activated killer cell-originated protein kinase is essential for chemoresistance against doxorubicin in cervical cancer cells. J. Biol. Chem. 2013, 288, 3585–3593.
- Park, J.H.; Jeong, Y.J.; Won, H.K.; Choi, S.Y.; Park, J.H.; Oh, S.M. Activation of TOPK by lipopolysaccharide promotes induction of inducible nitric oxide synthase through NF-kappaB activity in leukemia cells. Cell Signal. 2014, 26, 849–856.