Oxidative stress (OS) has a potential key role in the COVID-19 pathogenesis by triggering the NOD-like receptor family pyrin domain containing 3 inflammasome and nuclear factor-kB (NF-kB). While exposure to many pro-oxidants usually induces nuclear factor erythroid 2 p45-related factor2 (NRF2) activation and upregulation of antioxidant related elements expression, respiratory viral infections often inhibit NRF2 and/or activate NF-kB pathways, resulting in inflammation and oxidative injury. Hence, the use of radical scavengers like N-acetylcysteine and vitamin C, as well as of steroids and inflammasome inhibitors, has been proposed. The NRF2 pathway has been shown to be suppressed in severe SARS-CoV-2 patients. Pharmacological NRF2 inducers have been reported to inhibit SARS-CoV-2 replication, the inflammatory response, and transmembrane protease serine 2 activation, which for the entry of SARS-CoV-2 into the host cells through the angiotensin converting enzyme 2 receptor.
- SARS-CoV-2
- oxidative stress
- inflammation
- NRF2
- NF-kB
1. Introduction
The coronavirus disease 2019 (COVID-19) pandemic is caused by a novel severe acute respiratory syndrome (SARS)-like coronavirus (SARS-CoV-2) [1].
SARS-CoV-2 is an enveloped, non-segmented, positive sense RNA virus, widely distributed in humans and other mammals [2,3][2][3]. SARS-CoV-2 is dissimilar from the coronaviruses recognized to induce the ordinary cold, but it has been shown to have the same characteristics as the zoonotic SARS coronavirus (SARS-CoV) [4] and the Middle East respiratory syndrome (MERS) coronavirus [5]. Patients affected by COVID-19 often display no symptoms or mild symptoms (fever, cough, myalgia, and fatigue) and usually have a good prognosis. Many of these cases, however, progress to a more severe form of the illness, especially in older men experiencing other contemporary serious diseases [2,6,7,8][2][6][7][8]. Severe patients can suffer from symptoms correlated with lung [2[2][8][9],8,9], heart [8[8][10][11],10,11], kidney [8[8][12][13],12,13], neurological [14[14][15],15], gastrointestinal [16] and liver [9,16,17,18][9][16][17][18] injuries. Furthermore, there may be immune [9,12,19,20][9][12][19][20] and coagulation [21,22][21][22] impairment. Globally, as of December 27, 2020, there have been 79,232,555 confirmed COVID-19 cases, including 1,754,493 deaths [23].
Angiotensin converting enzyme 2 (ACE2) offers an access receptor for SARS-CoV-2 and SARS-CoV in humans by binding to the viral membrane spike (S) protein [24,25][24][25]. The quick recognition of ACE2 as SARS-CoV-2 receptor is mostly attributable to its recognition as the receptor for SARS-CoV about 17 years ago. In that case, ACE2 was recognized as the functional receptor for SARS-CoV after the fusion protein gene of SARS-CoV was reported [26]. By means of in vitro studies, Li et al. [27] found that: (1) ACE2 attached to the SARS-CoV S1 protein; (2) a soluble variety of ACE2, but not ACE1, inhibited the binding of the S1 protein with ACE2; (3) SARS-CoV reproduced in a very intense manner in ACE2-transfected, but not mock-transfected, cells. Furthermore, studies in vivo have clearly shown that ACE2 is a pivotal SARS-CoV receptor [28].
2. SARS-CoV-2 Cell Entry Mechanisms
2.1. SARS-CoV-2 Structural Basis
Like SARS-CoV, SARS-CoV-2 has four principal structural proteins: spike (S), envelope (E), membrane (M) and nucleocapsid (N), together with several additional proteins [29,30][29][30] (Figure 1). The S glycoprotein is a transmembrane protein (molecular weight of about 150 kDa) found in the virus outer portion [31]. Like SARS-CoV, S protein occurs as a trimer, with three receptor-binding S1 heads being placed on top of a membrane fusion S2 stalk [31] (Figure 1). S1, which binds to the peptidase domain of ACE2, is called the receptor-binding domain (RBD), while S2 catalyzes the membrane fusion, thus releasing the genetic material into the cells [31]. The crystal structures of the RBD of the S protein of SARS-CoV-2, both non-complexed [32] (protein data bank code 6VXX, https://www.rcsb.org (accessed on 31 December 2020)) or complexed with human ACE2 [33] (protein data bank code 6M0J, https://www.rcsb.org (accessed on 31 December 2020)) have been published previously. Recent studies, however, have established that there are slight differences between SARS-CoV-2 and SARS-CoV in receptor recognition [34]; these dissimilarities allow SARS-CoV-2 RBD to possess a slightly higher ACE2 receptor affinity than RBD of SARS-CoV [31], even though it results in being less accessible [32,35][32][35]. To retain its elevated infectivity despite a low accessibility, SARS-CoV-2 uses activation of host proteases, and this process crucially determines the infectivity and pathogenesis of SARS-CoV-2 infection [31]. In this context, it has previously been established that the pre-activation of furin, a host proprotein convertase [35[35][36],36], increases SARS-CoV-2 entrance into cells expressing ACE2 receptor by binding to a polybasic sequence motif at the S1/S2 border of the virus [31]. Furin-cleaved substrates then link to neuropilin-1 (NRP1), facilitating SARS-CoV-2 infectivity [36,37][36][37]. Moreover, transmembrane protease serine 2 (TMPRSS2) and lysosomal cathepsins, in addition to forcing SARS-CoV-2 entrance, have an additional impact with furin on SARS-CoV-2 entry [31]. Entered-SARS-CoV-2 will subsequently release its genomic material in the cytoplasm and be translated into the nuclei [38].
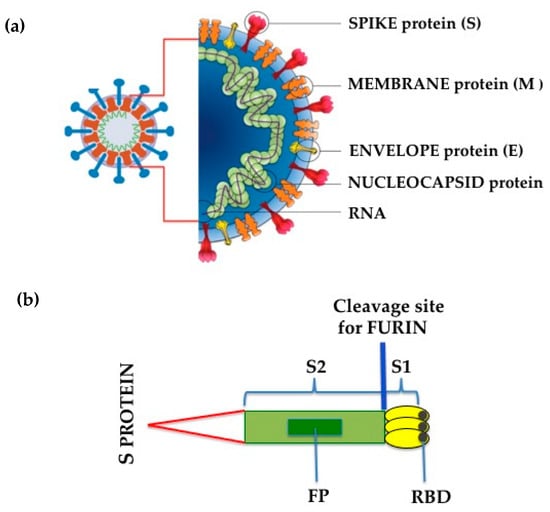
Figure 1. (a) SARS-CoV-2 structure; (b) Schematic drawing of SARS-CoV-2 Spike (S) protein. S1, receptor-binding subunit; S2, membrane fusion subunit; FP, fusion protein; RBD, receptor binding domain.
2.2. Structural Basis and Function of ACE2 Receptor
The renin-angiotensin system (RAS) plays a role in controlling blood volume and systemic vascular resistance, which at the same time affect cardiac output and arterial pressure [39]. ACE, a dipeptidyl carboxypeptidase in the RAS, converts the inactive angiotensin (Ang) I into the active and effective vasoconstrictor Ang II and inhibits the vasodilator Bradykinin [40]. ACE2 counterbalances ACE to a great extent by converting Ang I into Ang 1–9, an inert variety of Ang. It can also break down and hydrolyze the vasoconstrictor Ang II, into Ang 1–7, which acts as a strong vasodilator [41]. The ACE2 crystal structure and RBD of the S protein of SARS-CoV-2 complexed with human ACE2 have previously been reported [33,42][33][42] (protein data bank codes 1R42 and 6M0J, respectively, https://www.rcsb.org (accessed on 31 December 2020)). As just reviewed [43,44][43][44], ACE2 has multiple crucial protecting roles against hypertension, cardiovascular and lung diseases, and diabetes mellitus. Furthermore, the control of gut dysbiosis and vascular permeability by ACE2 has come out as an intrinsic mechanism of pulmonary hypertension and diabetes-related cardiovascular complications [44].
Very recently, ACE2 has been garnering widespread interest as a functional SARS-CoV-2 and SARS-CoV virus receptor by binding to the viral S protein, in this way contributing to pathogenesis of SARS [11,24,25,27][11][24][25][27]. ACE2 is ubiquitously expressed, with the highest levels in the epithelial cells of the lung, kidney and cardiomyocytes [45], although there is no lack of discordant voices, mostly for lung tissue [46]. Furthermore, recent studies based on single-cell RNA-sequence (scRNA-seq) data analysis have reported that ACE2 is widespread in many organs, including the lungs, heart, esophagus, kidneys, bladder, ileum, oral mucosa, and, particularly in the case of type II alveolar cells, cardiomyocytes, kidney proximal tubule cells, ileum and esophagus epithelial cells, and bladder urothelial cells [47]. Thank to this diffuse presence, ACE2 is involved in virus infection and diffusion. In addition, it has previously been found that infection with SARS-CoV and SARS-CoV-2 causes ACE2 shedding with subsequent downregulation of surface ACE2 expression [28,48][28][48]. In this context, in a small group of severe COVID-19 patients, Ang II plasma concentration was found to be significantly higher than in healthy controls [49], strengthening the hypothesis of a direct link between tissue ACE2 downregulation with systemic RAS imbalance.
As shown in Figure 2, recent evidence has shown that ectodomain shedding of ACE2 is mediated by ADAM17 (a disintegrin and metalloproteinase17), which in turn is upregulated by endocytosed SARS-CoV-2 S proteins [50] and other mechanisms [51,52,53,54][51][52][53][54].
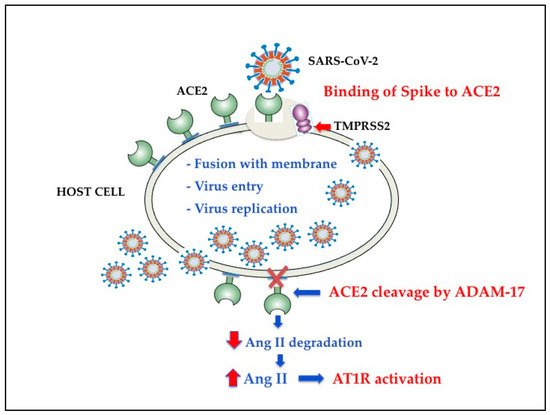
Figure 2. Schematic diagram of SARS-CoV-2 effects on renin angiotensin system. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; Ang II, Angiotensin II; Adam-17, a disintegrin and metalloproteinase17; AT1R, angiotensin II type-1 receptor; TMPRSS2, transmembrane protease serine 2.
The available body of facts indicates that Ang II binding to AT1R also controls the activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases [NOX] [54[54][55],55], one of the most important determinants of reactive oxygen species (ROS) generation. Hence, the SARS-CoV-2-induced ACE2 downregulation increases the binding of Ang II to AT1R, which, by triggering NOX, causes oxidative stress (OS) and inflammation in accordance with the COVID-19 severity [46].
3. Oxidative Stress and Inflammation Associated with SARS-CoV-2 Infection
3.1. Oxidative Stress (OS) in SARS-CoV-2 Infection
It is known that OS arises whenever there is an imbalance between ROS formation and antioxidant defenses. Alterations of the redox state towards oxidant conditions in infected cells is one of the key events in respiratory viral infections that is linked to inflammation and subsequent tissue damage [68,69,70][56][57][58]. Recent evidence indicates that OS play a crucial role also in COVID-19 infection [71,72,73,74,75][59][60][61][62][63]. Several in vitro and in vivo studies have shown that ROS overproduction induced by respiratory viruses is partially mediated by the activity of NOX (reviewed in [69][57]). As reported above, ACE2 shedding caused by SARS-CoV-2 fusion may be strictly related to RAS imbalance [43[43][47],47], and there is now evidence that Ang II controls NOX activation [54,55][54][55] (Figure 3). It has been suggested that NOX2 is a key event in killing bacteria and fungi, but it does not efficiently function against viruses [71][59]. In this regard, a recent study shows that OS induced by NOX2 activation is linked with severe clinical outcome and thrombotic events in COVID-19 patients [76][64]. ACE2 downregulation and OS are also associated with endothelial dysfunction via NOX activation and reduced availability of nitric oxide [77][65]. Furthermore, oxidized phospholipids (OxPLs), which are a product of OS and have been detected in the lungs of SARS-CoV patients [78][66], were found to be one of the main triggers of acute lung injury. As a matter of fact, OxPLs were shown to promote tissue factor expression [78][66], to activate endothelial cells to recruit monocytes [79[67][68],80], and to trigger macrophage activation through Nuclear Factor-κB (NF-kB) pathway [78][66]. It remains to be elucidated whether analogous pathways are also involved in SARS CoV-2 infection.
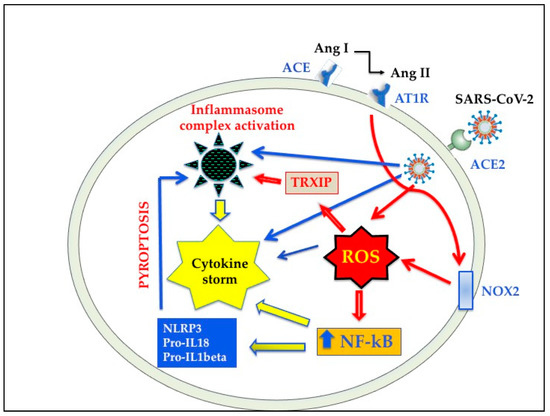
Oxidative stress and inflammation induced by SARS-CoV-2 infection. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; AT1R, angiotensin II type-1 receptor; NOX2, NADPH oxidase 2, NF-kB, nuclear factor kB; ROS, reactive oxygen species; TRXIP, thioredoxin interacting/inhibiting protein; NLRP3, NOD-like receptor protein 3.
It is well recognized that the levels of cellular free iron must be tightly regulated to avoid ROS generation via the Fenton reaction [81][69]. Upon SARS-CoV-2 infection, IL-6 in the cytokine storm increases ferritin and the production of hepcidin, which plays a main role in iron regulation. Since iron is sequestered by hepcidin in the enterocytes and macrophages, intracellular ferritin is augmented, leading to a reduced iron efflux from the cells. The stored iron may increase intracellular labile iron (II) pool and Fenton reaction, producing lipid ROS, and lead to ferroptosis, a novel form of regulated cell death [81][69]. In COVID-19 patients, the documented iron metabolism alterations may cause iron accumulation and overload, triggering ferroptosis in the cells of multiple organs [82,83][70][71].
Many lines of evidence show that viruses may also generate OS per se [69,70][57][58]. With regard to SARS-CoV, the viral protease 3CLpro has been previously shown to increase ROS generation in HL-CZ cells, with subsequent cell apoptosis and NF-kB-activation [84][72]. Another SARS-CoV protease, the 3a protein, has been linked with mitochondrial cell death pathway activation by triggering OS [69][57].
The mitochondrial respiratory chain is the main and most significant source of cellular ROS. However, while mitochondrial ROS production was once seen as merely an accidental by-product of oxygen metabolism of mitochondrial respiratory chain, it is now clear that ROS contribute to various signaling pathways [85][73]. Depending on the context and triggering stimuli, mitochondrial ROS production can lead to different cellular responses such as adaptation to hypoxia, differentiation, autophagy, inflammation, or to an immune response [86][74]. In general, viruses can modify mitochondrial dynamics in a highly specific manner so that they can successfully replicate [87][75]. Among the different mechanisms implicated, there are mitochondrial DNA damage, changes in mitochondrial membrane potential, variations in mitochondrial metabolic pathways and calcium homeostasis, modifications in number and distribution of mitochondria into the cells, weakening of antioxidant defense, and augmented OS [87,88][75][76]. Upon infection, viruses completely rely on host cell molecular machinery to survive and replicate [87,88][75][76]. Mitochondria defend host cells from SARS-CoV-2 virus through several mechanisms including cellular apoptosis, ROS production, autophagy, mitochondrial antiviral signaling system (MAVS) activation, DNA-dependent immune activation, and other things [89][77]. Current knowledge of how SARS-CoV-2 infection affects mitochondria and their ROS generation is limited. A prior study on SARS-CoV [90][78] showed that open reading frame-9b (Orf9b), one of the accessory proteins of the virus [91][79], alters host cell mitochondria morphology, disrupts MAVS, inhibits interferon (IFN) production and enhances autophagy, a cellular mechanism activated by ROS [92][80]. Consistent with the findings of SARS-CoV, Gordon et al. [93][81] recently reported that SARS-CoV-2 Orf9b interacts with mitochondrial translocase of outer membrane (TOM)70, although the functional consequences of this association were not examined. Very recently, Jiang et al. [94][82] reported that SARS-CoV-2 Orf9b localizes to the membrane of mitochondria and suppresses IFN-I response through association with TOM70. The altered activity of TOM70, by reducing constitutive calcium transfer to mitochondria, dampens mitochondrial respiration, affects cell bioenergetics, and induces autophagy [95][83].
During viral infections beyond an over-production of ROS, there is a decreased antioxidant defense, mainly Glutathione (GSH) depletion, in the host cells that directly or indirectly favor viral replication [96][84]. GSH, a tripeptide consisting of cysteine, glycine, and glutamate, is the main intracellular antioxidant that applies an efficient buffering role against ROS, through the thiol group of its cysteine which oxidizes to the disulfide form, then reduced back to the thiol form by glutathione reductase [70][58].
It has a principal role in cellular signaling and processes, as well as innate immune response to viruses [70][58].
A significant elevation in blood serum GSH reductase, derived from OS imbalance, was found in COVID-19 patients, especially when admitted to the intensive care unit [97][85]. Additionally, mounting evidence supports the concept that the reduced levels of GSH may underlie the COVID-19 severe clinical outcome and death [98][86].
3.2. Cross Talks between Oxidative Stress and Inflammation in SARS-CoV-2 Infection
Several studies have demonstrated that SARS-CoV-2 infection and the destruction of lung cells causes a local immune response, recruiting macrophages and monocytes that reply to the infection, release cytokines and prime adaptive T and B cell immune responses. In most patients, this process overcomes the infection. However, sometimes, a dysfunctional immune response occurs, which leads to a cytokine storm that mediates general lung inflammation [2,99,100][2][87][88]. Increased plasma concentrations of inflammatory markers such as C-reactive protein and ferritin, of many cytokines such as TNF-alpha, IL-1beta, IL-6 and IL-8, and chemokines such as MCP1, together with increased neutrophils/lymphocytes ratio [11[11][87][88],99,100], have been associated with gravity of SARS-CoV-2 infection and death [2,19,101][2][19][89].
SARS-CoV-2 infection in type 2 alveolar and other cells activates NOD-like receptor protein 3 (NLRP3), an element of the innate immune system that acts as a pattern recognition receptor that recognizes damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) [102][90] and takes part in multiprotein complexes called inflammasomes, which bring together sensor proteins (like NLRP3) [103,104][91][92]. NLRP3 inflammasome is very often associated with cellular death by apoptosis and pyroptosis [105[93][94][95],106,107], an inflammatory form of programmed cell death [108][96] that releases large amounts of pro-inflammatory mediators [109][97]. Accumulating data have established a causal role between the pyroptosis of alveolar type 2, endothelial and immune cells and the progression of lung damage [110,111,112,113,114,115][98][99][100][101][102][103]. The contemporary activation of alveolar macrophages further produces large amounts of proinflammatory cytokines and chemokines [116,117][104][105], which activate endothelial cells [118[106][107],119], platelets [120,121][108][109] and neutrophils [122,123][110][111] generating platelet neutrophil complexes at endothelium surface [124,125][112][113]. This sequestration of platelet neutrophil complexes in the pulmonary vasculature is the prelude of a highly inflammatory and pro-coagulant situation, a state called immunothrombosis [119,126,127][107][114][115]. Convincing evidence shows that immunothrombosis is a pivotal determinant of micro-thrombi and micro-emboli generation in the alveolar capillary circulation [128[116][117],129], of fibrin deposition within the alveoli, and in some cases of disseminated intravascular coagulation [130,131,132][118][119][120]. Furthermore, the huge associated increase of activated neutrophils in lung interstitial tissue and alveoli [133][121] can discharge high levels of extremely cytotoxic neutrophil extracellular traps [133][121]. These events play a crucial part in determining intra-lung cytokine storm and the consequent tissue damage that is a peculiarity of ARDS, an inflammatory disease with pulmonary epithelial and capillary endothelial cells dysfunction, alveolar macrophages and neutrophils infiltration, cell death, and fibrosis [134][122].
While it is likely that lung and other tissue damages in SARS-CoV-2 infection are the results of multifactorial mechanisms, very recent studies indicate that ROS may play a major role in the initiation and progression of this inflammatory process [135,136][123][124]. In this context, it has been reported that OS triggers the NLRP3 inflammasome [137,138][125][126]. Although it is conceivable that other pathological pathways participate in NLRP3 induction [139[127][128][129],140,141], OS activates NLRP3 inflammasome through NF-kB and thioredoxin interacting/inhibiting protein [135,136,137,138,142,143][123][124][125][126][130][131] activation. In addition, NF-kB up-regulates IL-18 and IL-1beta expression, further increasing NLRP3 inflammasome [141[129][131][132],143,144], as shown in Figure 3. This OS-induced overactivation of NLRP3 inflammasome may play a key role in the pathogenesis of severe SARS-CoV-2 infection. In fact, when the innate response cannot clear the infection, the resulting NLRP3 hyperactivation is harmful, leading to perturbation of mitochondrial function, the release of DAMPS and mounting pyroptosis [102,103,104][90][91][92] determining virus propagation and massive destruction of affected tissues [145,146][133][134].
References
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Qamar, M.T.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARSCoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 4, 313–319.
- Chan, J.W.; Ng, C.K.; Chan, Y.H.; Mok, T.Y.; Lee, S.; Chu, S.Y.; Law, W.L.; Lee, M.P.; Li, P.C. Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome [SARS]. Thorax 2003, 58, 686–689.
- Badawi, A.; Ryoo, S.G. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus [MERS-CoV]: A systematic review and meta-analysis. Int. J. Infect. Dis. 2016, 49, 129–133.
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513.
- Liu, K.; Fang, Y.Y.; Deng, Y.; Liu, W.; Wang, M.F.; Ma, J.P.; Xiao, W.; Wang, Y.N.; Zhong, M.H.; Li, C.H.; et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin. Med. J. 2020, 133, 1025–1031.
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069.
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422.
- Han, H.; Xie, L.; Liu, R.; Yang, J.; Liu, F.; Wu, K.; Chen, L.; Hou, W.; Feng, Y.; Zhu, C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China. J. Med. Virol. 2020, 92, 819–823.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult in patients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 82, 1708–1720.
- Li, Z.; Wu, M.; Yao, J.; Guo, J.; Liao, X.; Song, S.; Li, J.; Duan, G.; Zhou, Y.; Wu, X.; et al. Caution on kidney dysfunctions of COVID-19 patients. medRxiv 2020.
- Mao, L.; Wang, M.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; Li, Y.; Jin, H.; et al. Neurologic manifestations of hospitalized patients with COVID-19 in Wuhan, China: A retrospective case series study. JAMA Neurol. 2020, 77, 683–690.
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22.
- Yao, X.H.; Li, T.Y.; He, Z.C.; Ping, Y.F.; Liu, H.W.; Yu, S.C.; Mou, H.M.; Wang, L.H.; Zhang, H.R.; Fu, W.J. A pathological report of three COVID-19 cases by minimally invasive autopsies. Zhonghua Bing Li Xue Za Zhi 2020, 8, 411–417.
- Xie, H.; Zhao, J.; Lian, N.; Lin, S.; Xie, Q.; Zhuo, H. Clinical characteristics of non-ICU hospitalized patients with coronavirus disease 2019 and liver injury: A Retrospective study. Liver Int. 2020, 40, 1321–1326.
- Zhang, Y.; Zheng, L.; Liu, L.; Zhao, M.; Xiao, J.; Zhao, Q. Liver impairment in COVID-19 patients: A retrospective analysis of 115 cases from a single center in Wuhan city, China. Liver Int. 2020, 40, 2095–2103.
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H. Clinical and immunologic features in severe and moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 30, 2620–2629.
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768.
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120.
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847.
- Coronavirus WHO. COVID-19. WHO. 2020. Available online: (accessed on 31 December 2020).
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J. Virol. 2020, 94, e00127-20.
- Kuhn, J.H.; Li, W.; Choe, H.; Farzan, M. Angiotensin-converting enzyme 2: A functional receptor for SARS coronavirus. Cell. Mol. Life Sci. 2004, 61, 2738–2743.
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454.
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 ACE2 in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269.
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing antibodies against SARS-CoV-2 and other human Coronaviruses. Trends Immunol. 2020, 41, 355–359.
- Shang, J.; Wana, Y.; Luoa, C.; Yea, G.; Genga, Q.; Auerbacha, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734.
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220.
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224.
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092.
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860.
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Deborah, K.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865.
- Chen, Y.; Liu, Q.; Guo, D.J. Emerging coronaviruses: Genome structure, replication, and pathogenesis. Med. Virol. 2020, 92, 418–423.
- Chappell, M.C. Biochemical evaluation of the renin-angiotensin system: Then good, bad, and absolute? Am. J. Physiol. 2016, 310, 137–152.
- Erdos, E.G. Conversion of angiotensin I to angiotensin II. Amer. J. Med. 1976, 60, 749–759.
- Tikellis, C.; Thomas, M.C. Angiotensin-Converting Enzyme 2 [ACE2] is a key modulator of the Renin Angiotensin System in health and disease. Int. J. Pept. 2012, 25, 62–94.
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Natalie, A. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007.
- Wang, K.; Gheblawi, M.; Oudit, G.Y. Angiotensin converting enzyme 2: A double-edged sword. Circulation 2020, 142, 426–428.
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474.
- Burrell, L.M.; Johnston, C.I.; Tikellis, C.; Cooper, M.E. ACE2, a new regulator of the renin-angiotensin system. Trends Endocrinol. Metab. 2004, 15, 166–169.
- Hikmet, F.; Méar, L.; Edvinsson, A.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610.
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192.
- Datta, P.K.; Liu, F.; Fischer, T.; Rappaport, J.; Qin, X. SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 2020, 10, 7448–7464.
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374.
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell. Cardiol. 2014, 66, 167–176.
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell. 2010, 37, 551–566.
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733.
- Bzowska, M.; Jura, N.; Lassak, A.; Black, R.A.; Bereta, J. Tumour necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur. J. Biochem. 2004, 271, 2808–2820.
- Zablocki, D.; Sadoshima, J. Angiotensin II and Oxidative Stress in the Failing Heart. Antioxid. Redox Signal. 2013, 19, 1095–1109.
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-Induced Production of Mitochondrial Reactive Oxygen Species: Potential Mechanisms and Relevance for Cardiovascular Disease. Antioxid. Redox Signal. 2013, 19, 1085–1094.
- Delgado-Roche, L.; Mesta, F. Oxidative stress as key player in severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020, 51, 384–387.
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A. Redox Biology of Respiratory viral infections. Viruses 2018, 10, 392.
- Checconi, P.; De Angelis, M.; Marcocci, M.E.; Fraternale, A.; Magnani, M.; Palamara, A.T.; Nencioni, L. Redox-modulating agents in the treatment of viral Infections. Int. J. Mol. Sci. 2020, 21, 4084.
- Cecchini, R.; Cecchini, A.L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 2020, 143, 101–102.
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362.
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and oxidative stress. Biochemistry 2020, 85, 1543–1553.
- Fernandes, I.G.; de Brito, C.A.; Dos Reis, V.M.S.; Sato, M.N.; Pereira, N.Z. SARS-CoV-2 and other respiratory viruses: What does oxidative Stress have to do with it? Oxid. Med. Cell. Longev. 2020, 2020, 8844280.
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A review. Protein J. 2020, 39, 644–656.
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in Covid-19. Redox Biol. 2020, 36, 101655.
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044.
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll- like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249.
- Berliner, J.A.; Watson, A.D. A role for oxidized phospholipids in atherosclerosis. N. Engl. J. Med. 2005, 353, 9–11.
- Owens, A.P.; Passam, F.H.; Antoniak, S.; Marshall, S.M.; McDaniel, A.L.; Rudel, L.; Williams, J.C.; Hubbard, B.K.; Dutton, J.A.; Wang, J.; et al. Monocyte tissue factor-dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J. Clin. Investig. 2012, 122, 558–568.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tan, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Yang, M.; Lai, C.L. SARS-CoV-2 infection: Can ferroptosis be a potential treatment target for multiple organ involvement. Cell Death Discov. 2020, 6, 130.
- Edeas, M.; Saleh, J.; Peyssonnaux, C. Iron: Innocent bystander or vicious culprit in COVID-19 pathogenesis? Int. J. Infect. Dis. 2020, 97, 303–305.
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380.
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806.
- Dunn, J.D.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Anand, S.K.; Tikoo, S.K. Viruses as modulators of mitochondrial functions. Adv. Virol. 2013, 20, 738794.
- de las Heras, N.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Efects of Vitamin D. Antioxidants 2020, 9, 897.
- Burtscher, J.; Cappellano, G.; Omori, A.; Koshiba, T.; Millet, G.P. Mitochondria: In the Cross fire of SARS-CoV-2 and immunity. Science 2020, 23, 101631.
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-CoV ORF-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089.
- McBride, R.; Fielding, B.C. The role of severe acute respiratory syndrome (SARS)-coronavirus accessory proteins in virus pathogenesis. Viruses 2013, 4, 2902–2923.
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Danielle, L. A SARS-CoV-2 protein interaction map reveals targets for drug-repurposing. Nature 2020, 583, 459–468.
- Jiang, H.; Zhang, H.; Meng, Q.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000.
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P. TOM70 sustains cell bioenergetics by promoting IP3R3-Mediated ER to mitochondria Ca(2+) transfer. Curr. Biol. 2018, 28, 369–382.
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal. 2011, 15, 593–606.
- Cao, M.; Zhang, D.; Wang, Y.; Lu, Y.; Zhu, X.; Li, Y.; Xue, H.; Lin, Y.; Zhang, M.; Sun, Y.; et al. Clinical features of patients infected with the 2019 novel coronavirus [COVID-19] in Shanghai, China. medRxiv 2020. preprint.
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624.
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943.
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848.
- Gong, J.; Dong, H.; Xia, Q.; Huang, Z.; Wang, D.; Zhao, Y.; Liu, W.; Tu, S.; Zhang, M.; Wang, Q.; et al. Correlation analysis between disease severity and inflammation-related parameters in patients with COVID-19 pneumonia. medRxiv 2020.
- Martinon, F. Detection of immune danger signals by NALP3. J. Leukoc. Biol. 2008, 83, 507–511.
- Siu, K.; Yuen, K.; Castano-Rodriguez, C.; Ye, Z.; Yeung, M.; Fung, S.; Yuan, S.; Chan, C.; Yuen, K.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Vander Heide, R.S. Pulmonary and cardiac pathology in Covid-19: The first autopsy series from New Orleans. medRxiv 2020.
- Tian, S.; Hu, W.; Niu, L.; Liu, H.; Xu, H.; Xiao, S.Y. Pulmonary pathology of early phase 2019 novel coronavirus [COVID-19] pneumonia in two patients with lung cancer. J. Thorac. Oncol. 2020, 15, 700–704.
- Yang, M. Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV Infection, SSRN. Electron. J. 2020, 3527420.
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114.
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916.
- Faust, H.; Mangalmurti, N.S. Collateral damage: Necroptosis in the development of lung injury. Am. J. Phys. Lung Cell. Mol. Phys. 2020, 318, 215–225.
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell death in the lung: The apoptosis-necroptosis axis. Annu. Rev. Physiol. 2019, 81, 375–402.
- Ueno, H.; Matsuda, T.; Hashimoto, S.; Amaya, F.; Kitamura, Y.; Tanaka, M.; Kobayashi, A.; Maruyama, I.; Yamada, S.; Hasegawa, N.; et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 170, 1310–1316.
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844.
- Mason, R.J. Pathogenesis of COVID-19 from a cell biologic perspective. Eur. Respir. J. 2020, 55, 2000607.
- Aberdein, J.D.; Cole, J.; Bewley, M.A.; Marriott, H.M.; Dockrell, D.H. Alveolar macrophages in pulmonary host defence the unrecognized role of apoptosis as a mechanism of intracellular bacterial killing. Clin. Exp. Immunol. 2013, 174, 193–202.
- Losa García, J.E.; Rodríguez, F.M.; Martín de Cabo, M.R.; García Salgado, M.J.; Losada, J.P.; Villarón, L.G.; López, A.J.; Arellano, J.L. Evaluation of inflammatory cytokine secretion by human alveolar macrophages. Mediat. Inflamm. 1999, 8, 43–51.
- Han, S.; Mallampalli, R.K. The acute respiratory distress syndrome: From mechanism to translation. J. Immunol. 2015, 194, 855–860.
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in acute respiratory distress syndrome: Cross talks between inflammation and coagulation. Respiration 2017, 93, 212–225.
- Haouari, M. Platelet oxidative stress and its relationship with cardiovascular diseases in type 2 diabetes mellitus patients. Curr. Med. Chem. 2019, 26, 4145–4165.
- Freedman, J.E. Oxidative stress and platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 11–16.
- Øynebråten, I.; Barois, N.; Bergeland, T.; Küchler, A.M.; Bakke, O.; Haraldsen, G. Oligomerized, filamentous surface presentation of RANTES/CCL5 on vascular endothelial cells. Sci. Rep. 2015, 6, 9261.
- Sonmez, O.; Sonmez, M. Role of platelets in immune system and inflammation. Porto Biomed. J. 2017, 2, 311–314.
- Finsterbusch, M.; Schrottmaier, W.; Kral-Pointner, J.B.; Salzmann, M.; Asinger, A. Measuring and interpreting platelet-leukocyte aggregates. Platelets 2018, 29, 677–685.
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nácher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238.
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Kimball, A.S.; Obi, A.T.; Diaz, J.A.; Henke, P.K. The emerging role of NETs in venous thrombosis and immunothrombosis. Front. Immunol. 2016, 7, 236.
- Pfeiler, S.; Massberg, S.; Engelmann, B. Biological basis and pathological relevance of microvascular thrombosis. Thromb. Res. 2014, 133, 35–37.
- Prabhakaran, P.; Ware, L.B.; White, K.E.; Cros, M.T.; Matthay, M.A.; Olman, M.A. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am. J. Phys. Lung Cell. Mol. Phys. 2003, 285, 20–28.
- Sapru, A.; Curley, M.A.Q.; Brady, S.; Matthay, M.A.; Flori, H. Elevated PAI-1 is associated with poor clinical outcomes in pediatric patients with acute lung Injury. Intens. Care Med. 2010, 36, 157–163.
- Xue, M.; Sun, Z.; Shao, M.; Yin, J.; Deng, Z.; Zhang, J.; Xing, L.; Yang, X.; Chen, B.L.; Dong, Z.; et al. Diagnostic and prognostic utility of tissue factor for severe sepsis and sepsis-induced acute lung injury. J. Transl. Med. 2015, 13, 172.
- Yadav, H.; Kor, D.J. Platelets in the pathogenesis of acute respiratory distress Syndrome. Amer. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, 915–923.
- Gando, S.; Otomo, Y. Local hemostasis, immunothrombosis, and systemic disseminated intravascular coagulation in trauma and traumatic shock. Crit. Care 2015, 19, 72.
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, e138999.
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, S.D. Covid-19 does not lead to a “typical” acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300.
- Long, Y.; Liu, X.; Tan, X.Z.; Jiang, C.X.; Chen, S.W.; Liang, G.N.; He, X.M.; Wu, J.; Chen, T.; Xu, Y. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118-induced pyroptosis in endothelial cells. Ecotoxicol. Environ. Saf. 2020, 189, 109937.
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129.
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619.
- Boaru, S.G.; Borkham-Kamphorst, E.; Van de Leur, E.; Lehnen, E.; Liedtke, C.; Weiskirchen, R. NLRP3 inflammasome expression is driven by NF-kB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 2015, 458, 700–706.
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140.
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729.
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Halban, P.A.; Ehses, J.A. Cytokine production by islets in health and diabetes: Cellular origin, regulation and function. Trends Endocrinol. Metab. 2010, 21, 261–267.
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-induced NF-kB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026.
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Curr. Opin. Immunol. 2018, 50, 32–38.
- Zhao, M.; Bai, M.; Ding, G.; Zhang, Y.; Huang, S.; Jia, Z.; Zhang, A. Angiotensin II stimulates the NLRP3 inflammasome to induce podocyte injury and mitochondrial dysfunction. Kidney Dis. 2018, 4, 83–94.
- Van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 inflammasome dysregulated. Front. Immunol. 2020, 11, 1580.