Alternative splicing (AS) is a critical post-transcriptional regulatory mechanism used by more than 95% of transcribed human genes and responsible for structural transcript variation and proteome diversity.
- pre-messenger RNA,alternative splicing
- cancer
- mutation
- signalling
- splice variant
- pre-messenger RNA
- alternative splicing
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112]
1. Introduction
In higher eukaryotes, the primary gene transcripts, also called precursor messenger RNAs (pre-mRNAs), undergo a finely tuned post-transcriptional regulatory process that removes the non-coding regions (introns) and splices together the coding sequences (exons), thus generating the mature mRNAs. This mechanism is designated as pre-mRNA splicing and is a critical step in gene expression. In addition, it is well known that the splicing patterns of a gene vary widely as result of the process of alternative splicing (AS) that differentially retains or excludes certain exons from the pre-mRNA transcript. Consequently, various combinations of exons from a single gene can produce a diversity of mRNA variants, which is determinant to structural transcript variation and proteome diversity [1] and can generate different protein isoforms with related, distinct, or even opposing functions [2,3][2][3]. Remarkably, AS is a widespread event affecting more than 95% of transcribed human genes, as suggested by data provided by whole transcriptome sequencing projects [2,4][2][4]. This complex and tightly regulated mechanism is shared across different tissues and developmental stages, and frequently dysregulated in various human diseases, including cancer [5]. This dysregulation was verified in various types of cancer through detection of aberrant splicing patterns in tumor tissues when compared to their normal counterparts by high-throughput sequencing techniques [6,7,8,9][6][7][8][9]. Additionally, accumulating evidence clearly supports that the aberrant splicing profiles found in cancer are contributing to neoplastic transformation, cancer progression, and therapy resistance [10,11][10][11]. Therefore, it is of utmost relevance to identify pathological splicing isoforms for the development of new effective biomarkers, as well as to clarify the mechanisms behind aberrant AS, thereby elucidating its impact on cancer and providing novel therapeutic strategies.
2. Alternative RNA splicing: an overview
Pre-mRNA splicing consists of a multistep process orchestrated by the spliceosome, a huge RNA/protein complex comprising five small nuclear ribonucleoproteins (snRNPs; U1, U2, U4, U5, and U6) and numerous associated proteins [12,13]. Briefly, the reaction initiates with the assembly of an initial spliceosome complex through recognition of critical consensus splice sites at the pre-mRNA transcript, as schematically represented in Figure 1A. It comprises a stepwise process that begins with the recruitment of U1 snRNP to the 5′ splice site. Then, the splicing factor 1 (SF1), U2 snRNP auxiliary factor 2 (U2AF2), and U2 snRNP auxiliary factor (U2AF) 2, and U2AF 1recognize the branch point site (BPS), the polypyrimidine (poly-Y) tract, and the AG dinucleotide of the 3′ splice site region, respectively. The occupancy of these three consensus sequences induces the association of U2 snRNP with the BPS, which is further stabilized by the U2 snRNP component SF3B1. Consequently, intronic recognition prompts the engagement of U4/U6/U5 tri-snRNP with the complex, and subsequent formation of a catalytically inactive complex. This leads to several conformational and compositional rearrangements of spliceosomal components, including the dissociation of U1 and U4 snRNPs, which in turn promotes the formation of the activated spliceosome that catalyzes the splicing reaction [14]. Transcripts from nearly all protein-coding genes undergo one or more types of AS, giving rise to different mRNAs that differ in transcript degradation or are translated into alternative protein isoforms in a cell type-, organ-, or tissue-specific manner [2,4,15]. In higher eukaryotes, among the currently known AS events represented in Figure 1B, the most common is exon skipping [16], accounting for approximately 40% of all AS events, in which a cassette exon is removed from the pre-mRNA together with its flaking introns. Besides this, switching between alternative 5′ and 3′ splice site positions, mutually exclusive splicing of adjacent exons and differential retention of introns are also important variations of AS (Figure 1B). Other types of AS events include the use of alternative transcription start sites and alternative polyadenylation.
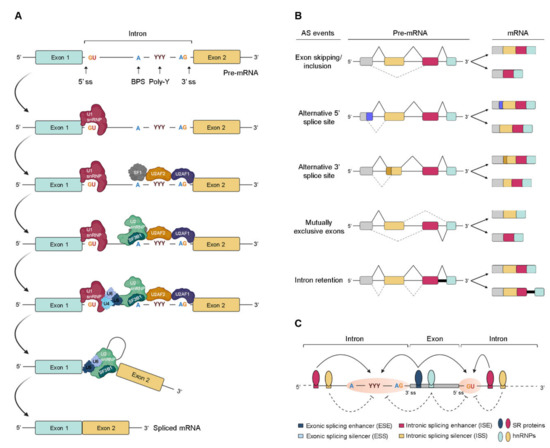
Figure 1. Regulation of pre-mRNA splicing. (A) Stepwise assembly of spliceosome on the pre-mRNA and catalysis of the splicing reaction to generate mature spliced mRNA. (B) Schematic representation of the most common alternative splicing AS events. The grey, yellow, red, and blue boxes represent different exons. The solid black and dotted grey lines indicate distinct splicing events. (C) Complex interplay between cis- and trans-acting factors in the regulation of AS. RNA-binding motif (RBM) proteins, serine/arginine-rich (SR) proteins, and heterogeneous (hn) ribonucleoproteins (hnRNPs) bind to exonic or intronic regulatory elements to promote or prevent the recognition of either 3′ or 5′ splice sites (ss) by the small nuclear (sn) RNPs (snRNPs) and splicing factors. The solid and dotted black arrows represent binding stimulation and inhibition, respectively; (ss—splice sites; BPS—branch point site; poly-Y—polypyrimidine tract; pre-mRNA—precursor messenger RNA; snRNPs—small nuclear ribonucleoprotein particle; SF1—splicing factor 1; U2AF—U2 snRNP auxiliary factor).
In AS, the regulated process consists of the recognition of an exon by the spliceosome. For this, splice site utilization is further regulated by cis-acting splicing-regulatory elements, which either promote or inhibit the use of adjacent splice sites by recruiting trans-acting splicing factors [17][12]. Thus, they are classified into exonic or intronic splicing enhancers (ESE/ISE) or silencers (ESS/ISS), depending on their positions and functions (Figure 1C). In general, enhancers are recognized by trans-acting factors belonging to the serine/arginine-rich (SR) protein family to facilitate splice site recognition and exon inclusion [18][13]. On the other hand, silencers usually interact with other types of trans-acting factors such as heterogeneous ribonucleoproteins (hnRNPs) to inhibit splice site recognition and promote exon skipping [2]. However, several AS events exist in which SR or hnRNP proteins act as inhibitors or enhancers of splicing, respectively.
3. Dysregulation of Alternative Splicing: the Case of Cancer
Cancer mainly evolves through successive genetic alterations and genomic dysregulation, but is also affected by the tumor microenvironment. These render oncogenes constitutively active and inactivate tumor-suppressor genes. As a result, cancer cells acquire specific abilities during tumor development, including self-sufficiency in growth signals, insensitivity to growth inhibitory signals, evasion of apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis [19][14]. These processes can also be dysregulated by AS, which in turn can generate variant proteins with altered physiological function [3]. Particularly, a recent systematic study performed by Kahles et al. reported that AS events are more frequent in cancer tissues compared to normal ones, and many of them are cancer-type specific [20][15]. Among the factors that can trigger aberrant AS, somatic mutations that disrupt splicing regulatory motifs, as well as mutations or expression changes in components of the core splicing machinery or splicing auxiliary factors, are frequently described [6,7,21,22,23,24][6][7][16][17][18][19].
Aberrant splicing in cancer has been widely linked to mutations creating cis-regulatory motifs that generate novel splice sites, as demonstrated by the discovery of almost 2000 splice site-creating mutations through a robust whole-exome analysis encompassing more than 8000 tumor samples across 33 cancer types [25][20]. One of the AS events frequently associated with these somatic mutations is intron retention, and mainly affects tumor suppressor genes such as TP53, ARID1A, and PTEN [7]. Importantly, most of the intron retention events are able to induce frameshifts in pre-mRNA sequence, resulting in the generation of premature termination codons (PTCs), which in turn leads to the degradation of the transcript through nonsense-mediated mRNA decay (NMD) or to the production of truncated proteins (e.g., dominant negative isoforms or neo-antigens). Interestingly, somatic exonic mutations have also been reported in oncogenes, particularly in ESE and ESS sequences [6], and associated with the generation of pro-tumorigenic variants.
Recurrent somatic mutations affecting the components of the early spliceosome complex formation have frequently been described in cancer, particularly in hematological malignancies, including myelodysplastic syndromes (MDS), other myeloid neoplasms, and chronic lymphocytic leukemia (CLL) [26,27,28][21][22][23]. Among the genes most affected by these mutations that almost always occur in a mutually exclusive manner are SF3B1 (splicing factor 3b subunit 1), SRSF2 (serine/arginine-rich splicing factor 2), U2AF1 (U2 small nuclear RNA auxiliary factor 1), and ZRSR2 (zinc finger RNA binding motif and serine/arginine rich 2) [26][21]. SF3B1, a subunit of the U2 snRNP that recognizes the BPS, is the most commonly mutated splicing regulator in numerous cancers, with a prevalence ranging from 5% in breast cancer to 81% in an MDS subtype [29][24]. Cancer-associated SF3B1 mutations are located within HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, Targets of rapamycin 1) domains, which are involved in protein–protein interactions and clustered in hotspots, namely K700, E622, R625, H662, and K666. Specifically, they are mainly related with the binding of SF3B1 to cryptic 3′ splice sites, located in regions with shorter and weaker poli-Y tracts, and consequently linked to aberrant BPS usage [22,30,31][17][25][26]. This abnormal assembly of spliceosome originates many mRNAs with a PTC, which are subsequently degraded by NMD.
Although the mechanism that induces the change of 3′ splice site usage by SF3B1 is not fully elucidated, it is hypothesized that these mutations alter the interaction of SF3B1 with other spliceosomal components required for BPS recognition. SRSF2 is a member of the SR protein family that binds to specific ESE sequences, namely CCNG or GGNG, through its RNA recognition motif (RRM) domain, and recruits U1 snRNP and U2AF to the 5′ and 3′ flanking splice sites, respectively [32][27]. This splicing regulator has also been found recurrently mutated, particularly in patients with MDS and chronic myelomonocytic leukemia (CMML) [26][21]. SRSF2 mutations predominantly occur at the P95 residue, which is located near the RRM domain [26][21]. According to several reports, these mutations change the RNA-binding affinity of SRSF2, favoring the recognition of C-rich CCNG over G-rich GGNG motifs in ESE consensus sites, which in turn leads to misregulation of exon inclusion [33,34][28][29]. The gene encoding UA2F1 is also mutated in myeloid malignancies, as well as in lung adenocarcinomas [26,35,36][21][30][31]. U2AF1 hotspot mutations occur almost exclusively at S34 and Q157 residues within the two conserved zinc-finger domains, thus affecting the recognition of the 3′ splice site AG motif [37,38][32][33]. In contrast to mutually exclusive hotspot mutations described for SF3B1, SRSF2, and U2AF1, ZRSR2 mutations are distributed throughout the gene and most are consistent with a loss-of-function phenotype [23][18]. In 2015, in addition to the major (or U2) spliceosome, ZRSR2 was also characterized as an essential component of the minor (or U12) spliceosome that catalyzes the processing of a distinct class of introns (U12-type introns). Particularly, it is involved in 3′ splice site recognition in U12 snRNA-dependent splicing, so that mutations in this gene are associated with an increase in the retention of U12-type introns [23][18].
Apart from genomic mutations, the pre-mRNA splicing of many genes related to cancer pathogenesis can also be disturbed by changes of the copy number or expression levels of splicing factors [39][34]. Actually, abnormal expression of several splicing factors have frequently been reported in solid tumors and closely associated with cancer development and progression, even in the absence of mutations [40,41,42,43][35][36][37][38]. One of the best characterized is the serine-arginine splicing factor 1 (SRSF1; formerly known as ASF or SF2), an SR protein involved in both constitutive and AS, as well as in other cellular processes. It is upregulated in several human tumors, including colon, breast, thyroid, small intestine, kidney, and lung, and its experimentally induced overexpression leads to the transformation of human and mouse mammary epithelial cells, suggesting that it acts as a proto-oncogene [44,45,46][39][40][41]. Until now, SRSF1 upregulation has been shown to affect many AS events in cancer-associated genes. In particular, SRSF1 overexpression induces an increase in the levels of oncogenic protein isoforms of RON [47][42], MNK2, and S6K1 [44][39] and of the anti-apoptotic isoforms Bcl-xL and MCL-1L [48][43], and a loss of the tumor suppressor isoform of BIN1 [44][39]. Curiously, the overexpression of hnRNP A1 and hnRNP A2/B1, two factors previously suggested to antagonize SR proteins, was also reported in lung, breast, and brain tumors [49,50,51,52][44][45][46][47]. Interestingly, in glioblastoma (GBM) cells, hnRNP A2/B1 showed splicing effects similar to the proto-oncogenic SR protein SRSF1 [52][47]. More recently, hnRNP A2 (as well as B1 and K) has been associated with enhanced expression of anti-apoptotic variants of BIN1 and CASP9, and decreased expression of the pro-apoptotic variant Bcl-xS [48][43], promoting the same phenotypic response as SRSF1 overexpression.
The major drivers of aberrant splicing profiles appear to be changes in the expression levels of splicing factors; however, the mechanisms behind the altered expression of the splicing factors in tumors are not yet fully understood. Although sporadic somatic mutations in genes encoding splicing factors have already been recurrently detected in solid tumors [43][38], it is widely recognized that oncogenic signaling has a central role [53][48]. Actually, abnormal activation of signaling pathways has been extensively reported in cancer. For instance, in colon cancer, oncogenic Kirsten rat sarcoma viral (KRAS) activates the RAS–MAPK pathway, leading to an increase in the expression levels of the AS factor polypyrimidine tract-binding protein 1 (PTBP1), activated via transcription factor ELK1. In turn, increased PTBP1 levels induce a shift in the AS of tumor-associated transcripts, namely, the small GTPase Ras-related C3 botulinum toxin substrate 1 (RAC1), adaptor protein NUMB, and PKM [54][49]. In addition to transcriptional stimulation of PTBP1 downstream of RAS, ERK was reported to phosphorylate the splicing factor SAM68, thereby inducing the binding of phospho-SAM68 to the 3′UTR of the SRSF1 transcript [55][50]. This binding promotes the retention of an intron required for production of full-length SRSF1 and prevents the downregulation of SRSF1 transcripts through the NMD pathway. Consequently, the increased SRSF1 levels, comparable in effect to the above described SRSF1 gene amplifications [44][39], induce a switch in AS of the RON gene transcripts, favoring the production of the oncogenic isoform RONΔex11. Phosphorylated SAM68 further stimulates inclusion of the variable exon 5 sequence into the CD44 mRNA, generating a pro-invasive cell adhesion protein variant [56][51].
Another MAPK pathway responds when cells experience physiologic stress. Osmotic stress triggers the MKK(3/6)-signaling cascade, leading to p38-activation, which upon nuclear translocation induces hnRNP A1 phosphorylation, followed by its export into the cytoplasm [57,58][52][53]. The corresponding decrease in nuclear splicing factor abundance is sufficient to change AS patterns. The PI3K/AKT signaling is another key pathway involved in cell survival and escape from apoptosis in numerous solid tumors. In non-small cell lung cancers (NSCLC), it was demonstrated that the activation of the PI3K/AKT pathway by oncogenic factors mediates the exclusion of the exon 3,4,5,6 cassette of CASP9 transcripts’ via the phosphorylation state of SRSF1, thus generating the anti-apoptotic Casp-9b isoform [59][54]. At the same time, AKT-mediated phosphorylation of hnRNPL induces its binding to a splice silencer element in Casp-9 pre-mRNA, further enhancing the exclusion of the exon cassette [60,61][55][56]. AKT activation also leads to phosphorylation and nuclear translocation of SR proteins, causing alternative exon inclusion in the fibronectin pre-mRNA [62][57]. Interestingly, in colorectal cells, inhibition of PI3K/AKT signaling led to increased expression of endogenous SRSF1, leading to the inclusion of an alternative exon, termed 3b, in the mRNA of the small GTPase RAC1, which generates the pro-tumorigenic splice variant RAC1B [63][58]. Later, it was described that SRPK1 and GSK3β act upstream of SRSF1, and are required to sustain RAC1B splicing in colorectal cancer (CRC) cells [64][59]. Particularly, it was shown that GSK3β indirectly regulates the levels of SRSF1 and RAC1B via SRPK1, since its depletion leads to a reduction of SRPK1 activity towards SRSF1, and a concomitant decrease in nuclear SRSF1 levels, resulting in less RAC1B generated. Another central hub of oncogenic signaling is the Wnt pathway, which is activated in many colorectal tumors. Remarkably, this pathway also modulates RAC1B splicing in CRC cells: It was described that the SRSF3 gene encoding splicing factor SRSF3/SRp20 is a transcriptional target for activated β-catenin/TCF4 complexes, leading to increased SRSF3 protein levels [65][60]. In a subsequent work, it was demonstrated that increased SRSF3 transcription following activation of the β-catenin/TCF4 pathway suppresses RAC1B splicing through SRSF3-mediated exclusion of exon 3b from the RAC1 mRNA [63][58]. Together, these examples show how signaling mechanisms affect alternative pre-mRNA splicing and change tumor-related gene expression.
3.1. Examples of Cancer-Associated Alternatively Spliced Variants
Several splice variants have been associated with different hallmarks of cancer, including initiation, progression, and metastasis. In Table 1, we highlight some of the most relevant AS events in cancer-associated genes involved in different steps of oncogenic transformation, as well as the types of cancer they are most often associated with. Other examples were listed in a recent review [66][61].
Table 1. Tumor-associated AS variants and the respective cancer-promoting process.
| Gene | Splicing Event | Biological Function | Cancer Types | References | |||||
|---|---|---|---|---|---|---|---|---|---|
| BCL2L1 | 5′ alternative splice site usage in exon 2 | Bcl-xL inhibits apoptosis | Lymphoma, glioma, breast, prostate, and liver cancer | [67,68,69,70,71] | [62][63][64][65][66] | ||||
| MKNK2 | Skipping of exon 14a and inclusion of exon 14b | MNK2b acts p38-MAPK-independent and promotes cell growth | Breast, colon, and lung cancer | [44,72,73] | [39][67][68] | ||||
| PKM | Skipping of exon 9 and inclusion of exon 10 | PKM2 stimulates aerobic glycolysis | Ovarian, gastric, liver, and colon cancer | [74,75,76,77] | [69][70][71][72] | ||||
| MST1R (RON) | Skipping of exon 11 | RONΔex11 induces cell motility and invasion | Colon, ovarian, brain, lung, and gastric cancer | [78,79,80,81,82] | [73][74][75][76][77] | ||||
| RPS6KB1 | Inclusion of three cassette exons 6a, 6b, and 6c with a PTC in exon 6c | RPS6KB1-2 promotes cell proliferation and tumor growth | Breast and lung cancer | [83, | [78 | 84] | ][79] | ||
| CCND1 | 5′ alternative splice site usage in exon 4 introduces a PTC | Cyclin D1b induces invasion and metastasis | Breast, lung, and prostate cancer | [85,86,87] | [80][81][82] | ||||
| VEGFA | Alternative 3′ splice site in exon 8 | VEGFA165 has pro-angiogenic activity | Colon, prostate, renal, and skin cancer | [88,89,90,91] | [83][84][85][86] | ||||
| CEACAM1 | Inclusion of exon 7 | CEACAM1-L accelerates metastasis progression | Colon cancer and metastatic melanoma | [92,93] | [87][88] | ||||
| CD44 | Inclusion of variable exon 6 | CD44-v6 induces migration and expression of mesenchymal markers | Colon cancer | [94,95,96] | [89][90][91] | ||||
| RAC1 | Inclusion of exon 3b | RAC1B increases cell survival and transformation | Colon, pancreas, thyroid, breast, and lung cancer | [63,97,98, | [ | 99,100, | 58 | 101,102] | ][92][93][94][95][96][97] |
| EGFR | Skipping of exon 4 | de4-EGFR promotes malignant transformation as constitutively active receptor variant | Glioma, prostate, and ovarian cancer | [103, | 98 | 104, | ] | 105] | [[99][100] |
| KLF6 | 5′ alternative splice site usage in exon 2 | KLF6-SV1 lacks nuclear localization and contributes to mesenchymal phenotype | Breast, lung, pancreatic, prostate, and liver cancer | [106,107,108,109,110] | [101][102][103][104][105] | ||||
| CTTN | Inclusion of exon 11 | Cortactin isoform-a increases cell migration | Colorectal cancer | [111] | [106] | ||||
| FAK | Deletion of exon 26 | The −26-exon FAK isoform is caspase-resistant and inhibits apoptosis | Breast cancer | [112] | [107] |
The listed genes are B-cell CLL/lymphoma 2-like 1 (BCL2L1), MAPK interacting serine/threonine kinase 2 (MKNK2), pyruvate kinase M (PKM), macrophage stimulating 1 receptor (MST1R), ribosomal protein S6 kinase B1 (RPS6KB1), cyclin D1 (CCND1), vascular endothelial growth factor A (VEGFA), CEA cell adhesion molecule 1 (CEACAM1), clusters of differentiation 44 (CD44), ras-related C3 botulinum toxin substrate 1 (RAC1), epidermal growth factor receptor (EGFR), Krüppel-like factor 6 (KLF6), cortactin (CTTN), and focal adhesion kinase (FAK); (PTC—premature termination codon).
References
- Blencowe, B. J., The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem Sci 2017, 42, (6), 407-408.Blencowe, B.J. The Relationship between Alternative Splicing and Proteomic Complexity. Trends Biochem. Sci. 2017, 42, 407–408.
- Wang, E. T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S. F.; Schroth, G. P.; Burge, C. B., Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, (7221), 470-6.Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476.
- Kim, H. K.; Pham, M. H. C.; Ko, K. S.; Rhee, B. D.; Han, J., Alternative splicing isoforms in health and disease. Pflugers Arch 2018, 470, (7), 995-1016.Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflug. Arch. 2018, 470, 995–1016.
- Pan, Q.; Shai, O.; Lee, L. J.; Frey, B. J.; Blencowe, B. J., Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008, 40, (12), 1413-5.Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415.
- Venables, J. P., Aberrant and alternative splicing in cancer. Cancer Res 2004, 64, (21), 7647-54.Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654.
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B., Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, (6), 1324-1335.Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335.
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y. J.; Park, W. Y.; Hong, D.; Park, P. J.; Lee, E., Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 2015, 47, (11), 1242-8.Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248.
- Chen, L.; Tovar-Corona, J. M.; Urrutia, A. O., Increased levels of noisy splicing in cancers, but not for oncogene-derived transcripts. Hum Mol Genet 2011, 20, (22), 4422-9.Chen, L.; Tovar-Corona, J.M.; Urrutia, A.O. Increased levels of noisy splicing in cancers, but not for oncogene-derived transcripts. Hum. Mol. Genet. 2011, 20, 4422–4429.
- Dvinge, H.; Bradley, R. K., Widespread intron retention diversifies most cancer transcriptomes. Genome Med 2015, 7, (1), 45.Dvinge, H.; Bradley, R.K. Widespread intron retention diversifies most cancer transcriptomes. Genome Med. 2015, 7, 45.
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R. K., RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 2016, 16, (7), 413-30.Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430.
- Paronetto, M. P.; Passacantilli, I.; Sette, C., Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ 2016, 23, (12), 1919-1929.Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell Death Differ. 2016, 23, 1919–1929.
- Rappsilber, J.; Ryder, U.; Lamond, A. I.; Mann, M., Large-scale proteomic analysis of the human spliceosome. Genome Res 2002, 12, (8), 1231-45.Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813.
- Jurica, M. S.; Moore, M. J., Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 2003, 12, (1), 5-14.Lee, Y.; Rio, D.C. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev. Biochem. 2015, 84, 291–323.
- Coltri, P. P.; Dos Santos, M. G. P.; da Silva, G. H. G., Splicing and cancer: Challenges and opportunities. Wiley Interdiscip Rev RNA 2019, 10, (3), e1527.Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Liu, Y.; Gonzalez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J. C.; Aebersold, R.; Venkitaraman, A. R.; Wickramasinghe, V. O., Impact of Alternative Splicing on the Human Proteome. Cell Rep 2017, 20, (5), 1229-1241.Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.
- Keren, H.; Lev-Maor, G.; Ast, G., Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 2010, 11, (5), 345-55.Bechara, E.G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733.
- Wang, Z.; Burge, C. B., Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, (5), 802-13.Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045.
- Lee, Y.; Rio, D. C., Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem 2015, 84, 291-323.Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042.
- Hanahan, D.; Weinberg, R. A., Hallmarks of cancer: the next generation. Cell 2011, 144, (5), 646-74.Zong, F.Y.; Fu, X.; Wei, W.J.; Luo, Y.G.; Heiner, M.; Cao, L.J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289.
- Kahles, A.; Lehmann, K. V.; Toussaint, N. C.; Huser, M.; Stark, S. G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research, N.; Ratsch, G., Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, (2), 211-224 e6.Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.
- Bechara, E. G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J., RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 2013, 52, (5), 720-33.Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69.
- Darman, R. B.; Seiler, M.; Agrawal, A. A.; Lim, K. H.; Peng, S.; Aird, D.; Bailey, S. L.; Bhavsar, E. B.; Chan, B.; Colla, S.; Corson, L.; Feala, J.; Fekkes, P.; Ichikawa, K.; Keaney, G. F.; Lee, L.; Kumar, P.; Kunii, K.; MacKenzie, C.; Matijevic, M.; Mizui, Y.; Myint, K.; Park, E. S.; Puyang, X.; Selvaraj, A.; Thomas, M. P.; Tsai, J.; Wang, J. Y.; Warmuth, M.; Yang, H.; Zhu, P.; Garcia-Manero, G.; Furman, R. R.; Yu, L.; Smith, P. G.; Buonamici, S., Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3' Splice Site Selection through Use of a Different Branch Point. Cell Rep 2015, 13, (5), 1033-45.Bejar, R.; Stevenson, K.E.; Caughey, B.A.; Abdel-Wahab, O.; Steensma, D.P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R.L.; Neuberg, D.; et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J. Clin. Oncol. 2012, 30, 3376–3382.
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; Miyano, S.; Thol, F.; Ganser, A.; Yang, H.; Haferlach, T.; Ogawa, S.; Koeffler, H. P., Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 2015, 6, 6042.Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome, C., Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627.
- Zong, F. Y.; Fu, X.; Wei, W. J.; Luo, Y. G.; Heiner, M.; Cao, L. J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; Hui, J., The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet 2014, 10, (4), e1004289.Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474.
- Jayasinghe, R. G.; Cao, S.; Gao, Q.; Wendl, M. C.; Vo, N. S.; Reynolds, S. M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; Varghese, R.; Huang, M.; Liang, W. W.; Wyczalkowski, M. A.; Sengupta, S.; Li, Z.; Payne, S. H.; Fenyo, D.; Miner, J. H.; Walter, M. J.; Cancer Genome Atlas Research, N.; Vincent, B.; Eyras, E.; Chen, K.; Shmulevich, I.; Chen, F.; Ding, L., Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep 2018, 23, (1), 270-281 e3.DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput. Biol. 2015, 11, e1004105.
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; Chalkidis, G.; Suzuki, Y.; Shiosaka, M.; Kawahata, R.; Yamaguchi, T.; Otsu, M.; Obara, N.; Sakata-Yanagimoto, M.; Ishiyama, K.; Mori, H.; Nolte, F.; Hofmann, W. K.; Miyawaki, S.; Sugano, S.; Haferlach, C.; Koeffler, H. P.; Shih, L. Y.; Haferlach, T.; Chiba, S.; Nakauchi, H.; Miyano, S.; Ogawa, S., Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, (7367), 64-9.Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615.
- Bejar, R.; Stevenson, K. E.; Caughey, B. A.; Abdel-Wahab, O.; Steensma, D. P.; Galili, N.; Raza, A.; Kantarjian, H.; Levine, R. L.; Neuberg, D.; Garcia-Manero, G.; Ebert, B. L., Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2012, 30, (27), 3376-82.Daubner, G.M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174.
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C. J.; Ellis, P.; Wedge, D. C.; Pellagatti, A.; Shlien, A.; Groves, M. J.; Forbes, S. A.; Raine, K.; Hinton, J.; Mudie, L. J.; McLaren, S.; Hardy, C.; Latimer, C.; Della Porta, M. G.; O'Meara, S.; Ambaglio, I.; Galli, A.; Butler, A. P.; Walldin, G.; Teague, J. W.; Quek, L.; Sternberg, A.; Gambacorti-Passerini, C.; Cross, N. C.; Green, A. R.; Boultwood, J.; Vyas, P.; Hellstrom-Lindberg, E.; Bowen, D.; Cazzola, M.; Stratton, M. R.; Campbell, P. J.; Chronic Myeloid Disorders Working Group of the International Cancer Genome, C., Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, (22), 3616-27; quiz 3699.Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630.
- Bonnal, S. C.; Lopez-Oreja, I.; Valcarcel, J., Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol 2020.Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, 4726–4734.
- DeBoever, C.; Ghia, E. M.; Shepard, P. J.; Rassenti, L.; Barrett, C. L.; Jepsen, K.; Jamieson, C. H.; Carson, D.; Kipps, T. J.; Frazer, K. A., Transcriptome sequencing reveals potential mechanism of cryptic 3' splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 2015, 11, (3), e1004105.Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; Dutertre, M.; Stern, M. H., Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 2016, 7, 10615.Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Cancer Genome Atlas Research, N.; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.
- Daubner, G. M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F. H., A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 2012, 31, (1), 162-74.Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26.
- Kim, E.; Ilagan, J. O.; Liang, Y.; Daubner, G. M.; Lee, S. C.; Ramakrishnan, A.; Li, Y.; Chung, Y. R.; Micol, J. B.; Murphy, M. E.; Cho, H.; Kim, M. K.; Zebari, A. S.; Aumann, S.; Park, C. Y.; Buonamici, S.; Smith, P. G.; Deeg, H. J.; Lobry, C.; Aifantis, I.; Modis, Y.; Allain, F. H.; Halene, S.; Bradley, R. K.; Abdel-Wahab, O., SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, (5), 617-30.Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016, 12, e1006384.
- Zhang, J.; Lieu, Y. K.; Ali, A. M.; Penson, A.; Reggio, K. S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J. L., Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A 2015, 112, (34), E4726-34.Zhang, J.; Manley, J.L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013, 3, 1228–1237.
- Kandoth, C.; McLellan, M. D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J. F.; Wyczalkowski, M. A.; Leiserson, M. D. M.; Miller, C. A.; Welch, J. S.; Walter, M. J.; Wendl, M. C.; Ley, T. J.; Wilson, R. K.; Raphael, B. J.; Ding, L., Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, (7471), 333-339.Ghigna, C.; Valacca, C.; Biamonti, G. Alternative splicing and tumor progression. Curr. Genom. 2008, 9, 556–570.
- Seiler, M.; Peng, S.; Agrawal, A. A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P. G.; Cancer Genome Atlas Research, N.; Buonamici, S.; Yu, L., Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 2018, 23, (1), 282-296 e4.Ghigna, C.; Moroni, M.; Porta, C.; Riva, S.; Biamonti, G. Altered expression of heterogenous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res. 1998, 58, 5818–5824.
- Ilagan, J. O.; Ramakrishnan, A.; Hayes, B.; Murphy, M. E.; Zebari, A. S.; Bradley, P.; Bradley, R. K., U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 2015, 25, (1), 14-26.Grosso, A.R.; Martins, S.; Carmo-Fonseca, M. The emerging role of splicing factors in cancer. EMBO Rep. 2008, 9, 1087–1093.
- Fei, D. L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C. L.; Bradley, R. K.; Varmus, H., Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet 2016, 12, (10), e1006384.Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427.
- Zhang, J.; Manley, J. L., Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 2013, 3, (11), 1228-37.Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193.
- Ghigna, C.; Valacca, C.; Biamonti, G., Alternative splicing and tumor progression. Curr Genomics 2008, 9, (8), 556-70.Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228.
- Ghigna, C.; Moroni, M.; Porta, C.; Riva, S.; Biamonti, G., Altered expression of heterogenous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Res 1998, 58, (24), 5818-24.Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117.
- Grosso, A. R.; Martins, S.; Carmo-Fonseca, M., The emerging role of splicing factors in cancer. EMBO Rep 2008, 9, (11), 1087-93.Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890.
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R. A.; Skotheim, R. I., Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, (19), 2413-27.Kedzierska, H.; Piekielko-Witkowska, A. Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett. 2017, 396, 53–65.
- Karni, R.; de Stanchina, E.; Lowe, S. W.; Sinha, R.; Mu, D.; Krainer, A. R., The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 2007, 14, (3), 185-93.Fielding, P.; Turnbull, L.; Prime, W.; Walshaw, M.; Field, J.K. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: A clinical marker of early lung cancer detection. Clin. Cancer Res. 1999, 5, 4048–4052.
- Anczukow, O.; Rosenberg, A. Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S. K.; Krainer, A. R., The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 2012, 19, (2), 220-8.Zhou, J.; Nong, L.; Wloch, M.; Cantor, A.; Mulshine, J.L.; Tockman, M.S. Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 2001, 34, 341–350.
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N. H.; Raimer, A.; Sun, S.; Jensen, M. A.; Hua, Y.; Allain, F. H.; Krainer, A. R., SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell 2015, 60, (1), 105-17.Zhou, J.; Allred, D.C.; Avis, I.; Martinez, A.; Vos, M.D.; Smith, L.; Treston, A.M.; Mulshine, J.L. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 2001, 66, 217–224.
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P. M.; Green, M. R.; Riva, S.; Biamonti, G., Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005, 20, (6), 881-90.Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472.
- Kedzierska, H.; Piekielko-Witkowska, A., Splicing factors of SR and hnRNP families as regulators of apoptosis in cancer. Cancer Lett 2017, 396, 53-65.Goncalves, V.; Pereira, J.F.S.; Jordan, P. Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes 2017, 9, 9.
- Fielding, P.; Turnbull, L.; Prime, W.; Walshaw, M.; Field, J. K., Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin Cancer Res 1999, 5, (12), 4048-52.Hollander, D.; Donyo, M.; Atias, N.; Mekahel, K.; Melamed, Z.; Yannai, S.; Lev-Maor, G.; Shilo, A.; Schwartz, S.; Barshack, I.; et al. A network-based analysis of colon cancer splicing changes reveals a tumorigenesis-favoring regulatory pathway emanating from ELK1. Genome Res. 2016, 26, 541–553.
- Zhou, J.; Nong, L.; Wloch, M.; Cantor, A.; Mulshine, J. L.; Tockman, M. S., Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 2001, 34, (3), 341-50.Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99.
- Zhou, J.; Allred, D. C.; Avis, I.; Martinez, A.; Vos, M. D.; Smith, L.; Treston, A. M.; Mulshine, J. L., Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat 2001, 66, (3), 217-24.Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695.
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S. S.; Bakacs, A.; Coppola, L.; Karni, R., Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 2011, 71, (13), 4464-72.van der Houven van Oordt, W.; Diaz-Meco, M.T.; Lozano, J.; Krainer, A.R.; Moscat, J.; Caceres, J.F. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J. Cell Biol. 2000, 149, 307–316.
- Goncalves, V.; Pereira, J. F. S.; Jordan, P., Signaling Pathways Driving Aberrant Splicing in Cancer Cells. Genes (Basel) 2017, 9, (1).Allemand, E.; Guil, S.; Myers, M.; Moscat, J.; Caceres, J.F.; Krainer, A.R. Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc. Natl. Acad. Sci. USA 2005, 102, 3605–3610.
- Hollander, D.; Donyo, M.; Atias, N.; Mekahel, K.; Melamed, Z.; Yannai, S.; Lev-Maor, G.; Shilo, A.; Schwartz, S.; Barshack, I.; Sharan, R.; Ast, G., A network-based analysis of colon cancer splicing changes reveals a tumorigenesis-favoring regulatory pathway emanating from ELK1. Genome Res 2016, 26, (4), 541-53.Shultz, J.C.; Goehe, R.W.; Wijesinghe, D.S.; Murudkar, C.; Hawkins, A.J.; Shay, J.W.; Minna, J.D.; Chalfant, C.E. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res. 2010, 70, 9185–9196.
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F. E.; Sette, C.; Ghigna, C.; Biamonti, G., Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 2010, 191, (1), 87-99.Goehe, R.W.; Shultz, J.C.; Murudkar, C.; Usanovic, S.; Lamour, N.F.; Massey, D.H.; Zhang, L.; Camidge, D.R.; Shay, J.W.; Minna, J.D.; et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Investig. 2010, 120, 3923–3939.
- Matter, N.; Herrlich, P.; Konig, H., Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, (6916), 691-5.Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584.
- van der Houven van Oordt, W.; Diaz-Meco, M. T.; Lozano, J.; Krainer, A. R.; Moscat, J.; Caceres, J. F., The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol 2000, 149, (2), 307-16.Blaustein, M.; Pelisch, F.; Tanos, T.; Munoz, M.J.; Wengier, D.; Quadrana, L.; Sanford, J.R.; Muschietti, J.P.; Kornblihtt, A.R.; Caceres, J.F.; et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat. Struct. Mol. Biol. 2005, 12, 1037–1044.
- Allemand, E.; Guil, S.; Myers, M.; Moscat, J.; Caceres, J. F.; Krainer, A. R., Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc Natl Acad Sci U S A 2005, 102, (10), 3605-10.Goncalves, V.; Matos, P.; Jordan, P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum. Mol. Genet. 2009, 18, 3696–3707.
- Shultz, J. C.; Goehe, R. W.; Wijesinghe, D. S.; Murudkar, C.; Hawkins, A. J.; Shay, J. W.; Minna, J. D.; Chalfant, C. E., Alternative splicing of caspase 9 is modulated by the phosphoinositide 3-kinase/Akt pathway via phosphorylation of SRp30a. Cancer Res 2010, 70, (22), 9185-96.Goncalves, V.; Henriques, A.F.; Pereira, J.F.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, 474–482.
- Goehe, R. W.; Shultz, J. C.; Murudkar, C.; Usanovic, S.; Lamour, N. F.; Massey, D. H.; Zhang, L.; Camidge, D. R.; Shay, J. W.; Minna, J. D.; Chalfant, C. E., hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest 2010, 120, (11), 3923-39.Goncalves, V.; Matos, P.; Jordan, P. The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA 2008, 14, 2538–2549.
- Vu, N. T.; Park, M. A.; Shultz, J. C.; Goehe, R. W.; Hoeferlin, L. A.; Shultz, M. D.; Smith, S. A.; Lynch, K. W.; Chalfant, C. E., hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J Biol Chem 2013, 288, (12), 8575-84.El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80.
- Blaustein, M.; Pelisch, F.; Tanos, T.; Munoz, M. J.; Wengier, D.; Quadrana, L.; Sanford, J. R.; Muschietti, J. P.; Kornblihtt, A. R.; Caceres, J. F.; Coso, O. A.; Srebrow, A., Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 2005, 12, (12), 1037-44.Xerri, L.; Hassoun, J.; Devilard, E.; Birnbaum, D.; Birg, F. BCL-X and the apoptotic machinery of lymphoma cells. Leuk. Lymphoma 1998, 28, 451–458.
- Goncalves, V.; Matos, P.; Jordan, P., Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum Mol Genet 2009, 18, (19), 3696-707.Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol. Rep. 2016, 35, 1013–1019.
- Goncalves, V.; Henriques, A. F.; Pereira, J. F.; Neves Costa, A.; Moyer, M. P.; Moita, L. F.; Gama-Carvalho, M.; Matos, P.; Jordan, P., Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 2014, 20, (4), 474-82.Olopade, O.I.; Adeyanju, M.O.; Safa, A.R.; Hagos, F.; Mick, R.; Thompson, C.B.; Recant, W.M. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J. Sci. Am. 1997, 3, 230–237.
- Goncalves, V.; Matos, P.; Jordan, P., The beta-catenin/TCF4 pathway modifies alternative splicing through modulation of SRp20 expression. RNA 2008, 14, (12), 2538-49.Mercatante, D.R.; Mohler, J.L.; Kole, R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J. Biol. Chem. 2002, 277, 49374–49382.
- El Marabti, E.; Younis, I., The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front Mol Biosci 2018, 5, 80.Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61.
- Xerri, L.; Hassoun, J.; Devilard, E.; Birnbaum, D.; Birg, F., BCL-X and the apoptotic machinery of lymphoma cells. Leuk Lymphoma 1998, 28, (5-6), 451-8.Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639.
- Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y., Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol Rep 2016, 35, (2), 1013-9.Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; et al. Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep. 2014, 7, 501–513.
- Olopade, O. I.; Adeyanju, M. O.; Safa, A. R.; Hagos, F.; Mick, R.; Thompson, C. B.; Recant, W. M., Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am 1997, 3, (4), 230-7.Chao, T.K.; Huang, T.S.; Liao, Y.P.; Huang, R.L.; Su, P.H.; Shen, H.Y.; Lai, H.C.; Wang, Y.C. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS ONE 2017, 12, e0182166.
- Mercatante, D. R.; Mohler, J. L.; Kole, R., Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J Biol Chem 2002, 277, (51), 49374-82.Shiroki, T.; Yokoyama, M.; Tanuma, N.; Maejima, R.; Tamai, K.; Yamaguchi, K.; Oikawa, T.; Noguchi, T.; Miura, K.; Fujiya, T.; et al. Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer-specific metabolism. Cancer Sci. 2017, 108, 931–940.
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S. L.; Takahashi, H., Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, (1), 55-61.Liu, W.R.; Tian, M.X.; Yang, L.X.; Lin, Y.L.; Jin, L.; Ding, Z.B.; Shen, Y.H.; Peng, Y.F.; Gao, D.M.; Zhou, J.; et al. PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, 846–861.
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C. L.; Thorsen, K.; Orntoft, T. F.; Mu, D.; Karni, R., The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol 2013, 229, (4), 630-9.Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; et al. Significance of Polypyrimidine Tract-Binding Protein 1 Expression in Colorectal Cancer. Mol. Cancer Ther. 2015, 14, 1705–1716.
- Maimon, A.; Mogilevsky, M.; Shilo, A.; Golan-Gerstl, R.; Obiedat, A.; Ben-Hur, V.; Lebenthal-Loinger, I.; Stein, I.; Reich, R.; Beenstock, J.; Zehorai, E.; Andersen, C. L.; Thorsen, K.; Orntoft, T. F.; Davis, R. J.; Davidson, B.; Mu, D.; Karni, R., Mnk2 alternative splicing modulates the p38-MAPK pathway and impacts Ras-induced transformation. Cell Rep 2014, 7, (2), 501-513.Zhou, Y.Q.; He, C.; Chen, Y.Q.; Wang, D.; Wang, M.H. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: Generation of different splicing RON variants and their oncogenic potential. Oncogene 2003, 22, 186–197.
- Chao, T. K.; Huang, T. S.; Liao, Y. P.; Huang, R. L.; Su, P. H.; Shen, H. Y.; Lai, H. C.; Wang, Y. C., Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS One 2017, 12, (7), e0182166.Mayer, S.; Hirschfeld, M.; Jaeger, M.; Pies, S.; Iborra, S.; Erbes, T.; Stickeler, E. RON alternative splicing regulation in primary ovarian cancer. Oncol. Rep. 2015, 34, 423–430.
- Shiroki, T.; Yokoyama, M.; Tanuma, N.; Maejima, R.; Tamai, K.; Yamaguchi, K.; Oikawa, T.; Noguchi, T.; Miura, K.; Fujiya, T.; Shima, H.; Sato, I.; Murata-Kamiya, N.; Hatakeyama, M.; Iijima, K.; Shimosegawa, T.; Satoh, K., Enhanced expression of the M2 isoform of pyruvate kinase is involved in gastric cancer development by regulating cancer-specific metabolism. Cancer Sci 2017, 108, (5), 931-940.Eckerich, C.; Schulte, A.; Martens, T.; Zapf, S.; Westphal, M.; Lamszus, K. RON receptor tyrosine kinase in human gliomas: Expression, function, and identification of a novel soluble splice variant. J. Neurochem. 2009, 109, 969–980.
- Liu, W. R.; Tian, M. X.; Yang, L. X.; Lin, Y. L.; Jin, L.; Ding, Z. B.; Shen, Y. H.; Peng, Y. F.; Gao, D. M.; Zhou, J.; Qiu, S. J.; Dai, Z.; He, R.; Fan, J.; Shi, Y. H., PKM2 promotes metastasis by recruiting myeloid-derived suppressor cells and indicates poor prognosis for hepatocellular carcinoma. Oncotarget 2015, 6, (2), 846-61.Krishnaswamy, S.; Mohammed, A.K.; Tripathi, G.; Alokail, M.S.; Al-Daghri, N.M. Splice variants of the extracellular region of RON receptor tyrosine kinase in lung cancer cell lines identified by PCR and sequencing. BMC Cancer 2017, 17, 738.
- Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; Takemasa, I.; Mizushima, T.; Ishii, M.; Mimori, K.; Ishii, H.; Doki, Y.; Mori, M.; Yamamoto, H., Significance of Polypyrimidine Tract-Binding Protein 1 Expression in Colorectal Cancer. Mol Cancer Ther 2015, 14, (7), 1705-16.Collesi, C.; Santoro, M.M.; Gaudino, G.; Comoglio, P.M. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol. Cell. Biol. 1996, 16, 5518–5526.
- Zhou, Y. Q.; He, C.; Chen, Y. Q.; Wang, D.; Wang, M. H., Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene 2003, 22, (2), 186-97.Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115.
- Mayer, S.; Hirschfeld, M.; Jaeger, M.; Pies, S.; Iborra, S.; Erbes, T.; Stickeler, E., RON alternative splicing regulation in primary ovarian cancer. Oncol Rep 2015, 34, (1), 423-30.Mei, H.; Wang, Y.; Fan, J.; Lin, Z. Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumour Biol. 2016, 37, 13369–13376.
- Eckerich, C.; Schulte, A.; Martens, T.; Zapf, S.; Westphal, M.; Lamszus, K., RON receptor tyrosine kinase in human gliomas: expression, function, and identification of a novel soluble splice variant. J Neurochem 2009, 109, (4), 969-80.Burd, C.J.; Petre, C.E.; Morey, L.M.; Wang, Y.; Revelo, M.P.; Haiman, C.A.; Lu, S.; Fenoglio-Preiser, C.M.; Li, J.; Knudsen, E.S.; et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 2190–2195.
- Krishnaswamy, S.; Mohammed, A. K.; Tripathi, G.; Alokail, M. S.; Al-Daghri, N. M., Splice variants of the extracellular region of RON receptor tyrosine kinase in lung cancer cell lines identified by PCR and sequencing. BMC Cancer 2017, 17, (1), 738.Li, R.; An, S.J.; Chen, Z.H.; Zhang, G.C.; Zhu, J.Q.; Nie, Q.; Xie, Z.; Guo, A.L.; Mok, T.S.; Wu, Y.L. Expression of cyclin D1 splice variants is differentially associated with outcome in non-small cell lung cancer patients. Hum. Pathol. 2008, 39, 1792–1801.
- Collesi, C.; Santoro, M. M.; Gaudino, G.; Comoglio, P. M., A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol 1996, 16, (10), 5518-26.Wang, Y.; Dean, J.L.; Millar, E.K.; Tran, T.H.; McNeil, C.M.; Burd, C.J.; Henshall, S.M.; Utama, F.E.; Witkiewicz, A.; Rui, H.; et al. Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res. 2008, 68, 5628–5638.
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R., S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep 2013, 3, (1), 103-15.Varey, A.H.; Rennel, E.S.; Qiu, Y.; Bevan, H.S.; Perrin, R.M.; Raffy, S.; Dixon, A.R.; Paraskeva, C.; Zaccheo, O.; Hassan, A.B.; et al. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: Balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br. J. Cancer 2008, 98, 1366–1379.
- Mei, H.; Wang, Y.; Fan, J.; Lin, Z., Alternative splicing of S6K1 promotes non-small cell lung cancer survival. Tumour Biol 2016, 37, (10), 13369-13376.Rennel, E.; Waine, E.; Guan, H.; Schuler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.; Bates, D.; et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br. J. Cancer 2008, 98, 1250–1257.
- Burd, C. J.; Petre, C. E.; Morey, L. M.; Wang, Y.; Revelo, M. P.; Haiman, C. A.; Lu, S.; Fenoglio-Preiser, C. M.; Li, J.; Knudsen, E. S.; Wong, J.; Knudsen, K. E., Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A 2006, 103, (7), 2190-5.Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131.
- Li, R.; An, S. J.; Chen, Z. H.; Zhang, G. C.; Zhu, J. Q.; Nie, Q.; Xie, Z.; Guo, A. L.; Mok, T. S.; Wu, Y. L., Expression of cyclin D1 splice variants is differentially associated with outcome in non-small cell lung cancer patients. Hum Pathol 2008, 39, (12), 1792-801.Pritchard-Jones, R.O.; Dunn, D.B.; Qiu, Y.; Varey, A.H.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230.
- Wang, Y.; Dean, J. L.; Millar, E. K.; Tran, T. H.; McNeil, C. M.; Burd, C. J.; Henshall, S. M.; Utama, F. E.; Witkiewicz, A.; Rui, H.; Sutherland, R. L.; Knudsen, K. E.; Knudsen, E. S., Cyclin D1b is aberrantly regulated in response to therapeutic challenge and promotes resistance to estrogen antagonists. Cancer Res 2008, 68, (14), 5628-38.Ieda, J.; Yokoyama, S.; Tamura, K.; Takifuji, K.; Hotta, T.; Matsuda, K.; Oku, Y.; Nasu, T.; Kiriyama, S.; Yamamoto, N.; et al. Re-expression of CEACAM1 long cytoplasmic domain isoform is associated with invasion and migration of colorectal cancer. Int. J. Cancer 2011, 129, 1351–1361.
- Varey, A. H.; Rennel, E. S.; Qiu, Y.; Bevan, H. S.; Perrin, R. M.; Raffy, S.; Dixon, A. R.; Paraskeva, C.; Zaccheo, O.; Hassan, A. B.; Harper, S. J.; Bates, D. O., VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer 2008, 98, (8), 1366-79.Ortenberg, R.; Galore-Haskel, G.; Greenberg, I.; Zamlin, B.; Sapoznik, S.; Greenberg, E.; Barshack, I.; Avivi, C.; Feiler, Y.; Zan-Bar, I.; et al. CEACAM1 promotes melanoma cell growth through Sox-2. Neoplasia 2014, 16, 451–460.
- Rennel, E.; Waine, E.; Guan, H.; Schuler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.; Bates, D.; Harper, S., The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br J Cancer 2008, 98, (7), 1250-7.Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356.
- Bates, D. O.; Cui, T. G.; Doughty, J. M.; Winkler, M.; Sugiono, M.; Shields, J. D.; Peat, D.; Gillatt, D.; Harper, S. J., VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res 2002, 62, (14), 4123-31.Saito, S.; Okabe, H.; Watanabe, M.; Ishimoto, T.; Iwatsuki, M.; Baba, Y.; Tanaka, Y.; Kurashige, J.; Miyamoto, Y.; Baba, H. CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol. Rep. 2013, 29, 1570–1578.
- Pritchard-Jones, R. O.; Dunn, D. B.; Qiu, Y.; Varey, A. H.; Orlando, A.; Rigby, H.; Harper, S. J.; Bates, D. O., Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br J Cancer 2007, 97, (2), 223-30.Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl. Med. 2015, 4, 1033–1043.
- Ieda, J.; Yokoyama, S.; Tamura, K.; Takifuji, K.; Hotta, T.; Matsuda, K.; Oku, Y.; Nasu, T.; Kiriyama, S.; Yamamoto, N.; Nakamura, Y.; Shively, J. E.; Yamaue, H., Re-expression of CEACAM1 long cytoplasmic domain isoform is associated with invasion and migration of colorectal cancer. Int J Cancer 2011, 129, (6), 1351-61.Singh, A.; Karnoub, A.E.; Palmby, T.R.; Lengyel, E.; Sondek, J.; Der, C.J. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, 9369–9380.
- Ortenberg, R.; Galore-Haskel, G.; Greenberg, I.; Zamlin, B.; Sapoznik, S.; Greenberg, E.; Barshack, I.; Avivi, C.; Feiler, Y.; Zan-Bar, I.; Besser, M. J.; Azizi, E.; Eitan, F.; Schachter, J.; Markel, G., CEACAM1 promotes melanoma cell growth through Sox-2. Neoplasia 2014, 16, (5), 451-60.Pelisch, F.; Khauv, D.; Risso, G.; Stallings-Mann, M.; Blaustein, M.; Quadrana, L.; Radisky, D.C.; Srebrow, A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J. Cell Biochem. 2012, 113, 2319–2329.
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G.; Gulotta, G.; Dieli, F.; De Maria, R.; Stassi, G., CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, (3), 342-56.Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21.
- Saito, S.; Okabe, H.; Watanabe, M.; Ishimoto, T.; Iwatsuki, M.; Baba, Y.; Tanaka, Y.; Kurashige, J.; Miyamoto, Y.; Baba, H., CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol Rep 2013, 29, (4), 1570-8.Matos, P.; Jordan, P. Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 2008, 6, 1178–1184.
- Yan, Y.; Zuo, X.; Wei, D., Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med 2015, 4, (9), 1033-43.Faria, M.; Capinha, L.; Simoes-Pereira, J.; Bugalho, M.J.; Silva, A.L. Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int. J. Endocrinol. 2016, 2016, 1972367.
- Singh, A.; Karnoub, A. E.; Palmby, T. R.; Lengyel, E.; Sondek, J.; Der, C. J., Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004, 23, (58), 9369-80.Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B.A.; Bugalho, M.J.; Silva, A.L. RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS ONE 2017, 12, e0172689.
- Pelisch, F.; Khauv, D.; Risso, G.; Stallings-Mann, M.; Blaustein, M.; Quadrana, L.; Radisky, D. C.; Srebrow, A., Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem 2012, 113, (7), 2319-29.Wang, H.; Zhou, M.; Shi, B.; Zhang, Q.; Jiang, H.; Sun, Y.; Liu, J.; Zhou, K.; Yao, M.; Gu, J.; et al. Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia 2011, 13, 461–471.
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H., RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, (1).Wang, H.; Shi, B.; Zhang, Q.; Jiang, H.; Hu, S.; Kong, J.; Yao, M.; Yang, S.; Li, Z. Growth and metastasis suppression of glioma xenografts expressing exon 4-deletion variant of epidermal growth factor receptor by monoclonal antibody CH12-mediated receptor degradation. FASEB J. 2012, 26, 73–80.
- Matos, P.; Jordan, P., Increased Rac1b expression sustains colorectal tumor cell survival. Mol Cancer Res 2008, 6, (7), 1178-84.Zhang, P.; Zhang, P.; Zhou, M.; Jiang, H.; Zhang, H.; Shi, B.; Pan, X.; Gao, H.; Sun, H.; Li, Z. Exon 4 deletion variant of epidermal growth factor receptor enhances invasiveness and cisplatin resistance in epithelial ovarian cancer. Carcinogenesis 2013, 34, 2639–2646.
- Faria, M.; Capinha, L.; Simoes-Pereira, J.; Bugalho, M. J.; Silva, A. L., Extending the Impact of RAC1b Overexpression to Follicular Thyroid Carcinomas. Int J Endocrinol 2016, 2016, 1972367.Hatami, R.; Sieuwerts, A.M.; Izadmehr, S.; Yao, Z.; Qiao, R.F.; Papa, L.; Look, M.P.; Smid, M.; Ohlssen, J.; Levine, A.C.; et al. KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci. Transl. Med. 2013, 5, 169.
- Faria, M.; Matos, P.; Pereira, T.; Cabrera, R.; Cardoso, B. A.; Bugalho, M. J.; Silva, A. L., RAC1b overexpression stimulates proliferation and NF-kB-mediated anti-apoptotic signaling in thyroid cancer cells. PLoS One 2017, 12, (2), e0172689.DiFeo, A.; Feld, L.; Rodriguez, E.; Wang, C.; Beer, D.G.; Martignetti, J.A.; Narla, G. A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res. 2008, 68, 965–970.
- Wang, H.; Zhou, M.; Shi, B.; Zhang, Q.; Jiang, H.; Sun, Y.; Liu, J.; Zhou, K.; Yao, M.; Gu, J.; Yang, S.; Mao, Y.; Li, Z., Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia 2011, 13, (5), 461-71.Hartel, M.; Narla, G.; Wente, M.N.; Giese, N.A.; Martignoni, M.E.; Martignetti, J.A.; Friess, H.; Friedman, S.L. Increased alternative splicing of the KLF6 tumour suppressor gene correlates with prognosis and tumour grade in patients with pancreatic cancer. Eur. J. Cancer 2008, 44, 1895–1903.
- Wang, H.; Shi, B.; Zhang, Q.; Jiang, H.; Hu, S.; Kong, J.; Yao, M.; Yang, S.; Li, Z., Growth and metastasis suppression of glioma xenografts expressing exon 4-deletion variant of epidermal growth factor receptor by monoclonal antibody CH12-mediated receptor degradation. FASEB J 2012, 26, (1), 73-80.Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H.L.; Qiao, R.F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005, 65, 5761–5768.
- Zhang, P.; Zhang, P.; Zhou, M.; Jiang, H.; Zhang, H.; Shi, B.; Pan, X.; Gao, H.; Sun, H.; Li, Z., Exon 4 deletion variant of epidermal growth factor receptor enhances invasiveness and cisplatin resistance in epithelial ovarian cancer. Carcinogenesis 2013, 34, (11), 2639-46.Yea, S.; Narla, G.; Zhao, X.; Garg, R.; Tal-Kremer, S.; Hod, E.; Villanueva, A.; Loke, J.; Tarocchi, M.; Akita, K.; et al. Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology 2008, 134, 1521–1531.
- Hatami, R.; Sieuwerts, A. M.; Izadmehr, S.; Yao, Z.; Qiao, R. F.; Papa, L.; Look, M. P.; Smid, M.; Ohlssen, J.; Levine, A. C.; Germain, D.; Burstein, D.; Kirschenbaum, A.; DiFeo, A.; Foekens, J. A.; Narla, G., KLF6-SV1 drives breast cancer metastasis and is associated with poor survival. Sci Transl Med 2013, 5, (169), 169ra12.Wang, Z.N.; Liu, D.; Yin, B.; Ju, W.Y.; Qiu, H.Z.; Xiao, Y.; Chen, Y.J.; Peng, X.Z.; Lu, C.M. High expression of PTBP1 promote invasion of colorectal cancer by alternative splicing of cortactin. Oncotarget 2017, 8, 36185–36202.
- DiFeo, A.; Feld, L.; Rodriguez, E.; Wang, C.; Beer, D. G.; Martignetti, J. A.; Narla, G., A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res 2008, 68, (4), 965-70.Yao, L.; Li, K.; Peng, W.; Lin, Q.; Li, S.; Hu, X.; Zheng, X.; Shao, Z. An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J. Transl. Med. 2014, 12, 136.
- Hartel, M.; Narla, G.; Wente, M. N.; Giese, N. A.; Martignoni, M. E.; Martignetti, J. A.; Friess, H.; Friedman, S. L., Increased alternative splicing of the KLF6 tumour suppressor gene correlates with prognosis and tumour grade in patients with pancreatic cancer. Eur J Cancer 2008, 44, (13), 1895-903.
- Narla, G.; DiFeo, A.; Yao, S.; Banno, A.; Hod, E.; Reeves, H. L.; Qiao, R. F.; Camacho-Vanegas, O.; Levine, A.; Kirschenbaum, A.; Chan, A. M.; Friedman, S. L.; Martignetti, J. A., Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res 2005, 65, (13), 5761-8.
- Yea, S.; Narla, G.; Zhao, X.; Garg, R.; Tal-Kremer, S.; Hod, E.; Villanueva, A.; Loke, J.; Tarocchi, M.; Akita, K.; Shirasawa, S.; Sasazuki, T.; Martignetti, J. A.; Llovet, J. M.; Friedman, S. L., Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology 2008, 134, (5), 1521-31.
- Wang, Z. N.; Liu, D.; Yin, B.; Ju, W. Y.; Qiu, H. Z.; Xiao, Y.; Chen, Y. J.; Peng, X. Z.; Lu, C. M., High expression of PTBP1 promote invasion of colorectal cancer by alternative splicing of cortactin. Oncotarget 2017, 8, (22), 36185-36202.
- Yao, L.; Li, K.; Peng, W.; Lin, Q.; Li, S.; Hu, X.; Zheng, X.; Shao, Z., An aberrant spliced transcript of focal adhesion kinase is exclusively expressed in human breast cancer. J Transl Med 2014, 12, 136.