Lipids are the major structural constituents of cell membranes.
- microbiome
- lipidomics
- metabolomics
- gut
- lipids
1. Lipids and Gut Microbes
Lipids are the major structural constituents of cell membranes. As energy storage molecules, they also store almost twice the energy as that liberated from protein or carbohydrate catabolism. Moreover, lipids regulate many essential biological functions, including intra-cellular signaling processes. For instance, sphingolipids (SPs), particularly ceramides, have a part to play in regulation of cell signaling and apoptosis [1]. Other lipids such as diacylglycerols (DGs) act as intermediates of energy metabolism and as signaling molecules [2]. Overall, lipid metabolism exhibits spatial and dynamic complexity at multiple levels. Thus, it is not surprising that lipid disturbances have important physiological consequences impacting human health [3].
The human gut, on the other hand, harbors metabolically-active microbial communities, which have profound impact on the absorption, digestion, metabolism and excretion of lipids [4]. There is a growing consensus that gut microbes and their metabolic activity alter the metabolic state of the host. Most recent studies in this field suggest the integration of the gut microbiome and metabolome, rather than sole focus on microbial taxonomic profiling, affords better understanding of microbiome-mediated (patho)physiological processes [5,6][5][6]. Thus, metabolome-based strategies for the study of gut microbial communities at both structural and functional levels are gaining much-warranted increasing attention.
Previous studies of the gut microbiome-metabolome co-axis have mainly focused on water-soluble, polar metabolites (e.g., tryptophan catabolites, amino acids, tricarboxylic acid cycle (TCA) intermediates), while the microbial-host-lipid co-axis has not received as much attention. In this review, we highlight recent fecal lipidomics studies aimed at deciphering the molecular lipids that are secreted, hydrolyzed or transformed by gut microbial communities. We also discuss mechanistic studies, involving integration of shotgun metagenomics and lipidomics data, and how these approaches may lead to better understanding of the impact of microbial lipids on host physiology. To this end, we highlight the emerging applications of genome-scale metabolic modeling (GSMM) as a way to study co-metabolism between the microbes as well as host–microbe interactions via lipidomes.
2. Lipid Pool of the Human Gut
A link between host molecular lipids, gut microbiota and human health is already evident, given the associations between them in several clinical studies [7,8][7][8]. Gut microbiota can both modulate the amount of energy that is extracted from food during digestion, and synthesize lipids and metabolites that may have an impact on human health. Alterations in the gut microbiota—host lipidome have been linked obesity and the development of obesity-related illnesses, among others. In humans, the diversity of the gut microbiome has been found to have a negative association with BMI and serum triacylglycerols (TGs) [9]. Liu et al. reported that gestational diabetes mellitus comorbidity was strongly associated with both a specific gut microbial composition and with the circulating lipidome [10]. Here, relative abundances of Faecalibacterium and Prevotella showed linkage with circulating lipids, in particular with lysophosphatidylethanolamine, and phosphatidylglycerols. Recently, Benítez–Páez et al. used a multi-omics approach (metabolomics, lipidomics and shotgun metagenomics) to characterize the impact of arabinoxylan oligosaccharides in overweight individuals [11]. They found that an arabinoxylan-enriched diet altered the host metabolic state, including ceramide and choline levels, which subsequently affected the abundance of Prevotella and Clostridial species in the gut. Animal studies also posit that gut microbes can modulate the host’s lipidome. For instance, Albouery et al. investigated how colonization of germ-free (GF) mice by the fecal microbiota of young or old donor mice impacted the lipid content of the brain and liver [12]. Mice receiving fecal bacteria from aged mice exhibited increased total monounsaturated fatty acids, and a reduction in the relative amounts of cholesterol and total polyunsaturated fatty acids in the brain cortex. In addition, the transfer of microbiota from aged to young mice modified the relative abundances of the different lipid classes and the fatty acid content of the liver. In another study, Just et al. studied how diets enriched with primary bile acids (BAs) with or without addition of lard or palm oil, impacted gut microbiota composition and function in mice [13]. The authors reported that the lard + BA-enriched diet increased the fat mass of colonized mice, but not in the GF mice, as compared to palm oil. Subsequently, these effects were associated with impaired glucose tolerance and elevated TGs, cholesteryl esters and monounsaturated fatty acids in the livers of the colonized mice.
From a clinical perspective, understanding the intricate relationship between systemic and microbial lipids may provide a unique avenue to study human metabolism. The fecal lipidome provides a non-invasive strategy to study the lipophilic activity of gut microbes, and their co-metabolism [14]. However, feces represent a highly heterogeneous sample matrix and, as such, pose several analytical challenges [15]. To extract useful information from stool samples, optimal sample preparation, handling, along with accurate and reliable analytical methods are required. Gregory et al. reported the lipidome composition of human stool samples for the first time [16]. The authors identified over 500 intact lipid species across six of eight different LIPID MAPS categories: glycerophospholipids, fatty acyls (FAs), SPs, glycerolipids, sterol lipids and prenol lipids [16]. Van Meulebroek et al. later optimized the lipidomics protocol for the analysis of stool samples to comprehensively map the lipidome [17]. More recently, Trošt et al. also screened fecal lipidome profiles from 10 healthy human volunteers [18]. The authors report that ceramides, DGs and TGs were the most abundant lipids found in stools. Lipids classes that are commonly detected at higher concentrations in plasma, e.g., lysophosphocholines, glycerophosphocholines and sphingomyelins were also present in the fecal samples, however, they were found at markedly lower concentrations [19].
One major challenge in fecal lipidome analysis is a largely technical one, i.e., the ability to identify and quantify the entire set of lipids in the stool. Processing of raw fecal lipidomics data shares common steps with mass spectrometric (MS)-based metabolomic analysis [20,21,22][20][21][22]. At present, identification of ‘unknown’ metabolites using tandem mass-spectrometry (MS/MS) is challenging, as limited number of reference spectra and/or authentic standards are available. Gao et al. [23] and Phua et al. [24] have identified metabolites from fecal samples using NIST MS libraries (www.chemdata.nist.gov). Several other commercial and non-commercial databases such as FiehnLib [25], Golm Metabolome Database [26], Human Metabolome Database (HMDB) [27], LIPID MAPS [28] and METLIN [29], have enabled identification of metabolites, including lipids. Additionally, in silico tools have also been developed to facilitate lipid identification, including structure database-dependent methods and spectra library-dependent methods. Spectra library independent tools such as LipidHunter, MS-DIAL and MZmine2 also provide specific workflows for profiling the lipidome. The integration of several metabolite databases might extend the coverage of lipids; however, this results in a markedly more complex workflow. CEU Mass Mediator [30] is a computationally-efficient integrative framework developed for this purpose; it can search for molecular lipids using multiple databases. However, to enable precise identification of lipids, both in silico as well as analytical advances are needed (reviewed in detail by Wei et al. 2019) [3].
Together, the fecal lipidome provides information regarding the metabolic interplay between the host, diet and gut microbiome [31]. An increasing number of studies suggest that the stool lipidome provides a functional readout of microbial metabolism; however, quantification of the lipids in the feces will likely result in a combination of host-originating, microbe-originating or host-microbiome co-metabolic products. Thus, whilst the fecal lipidome can suggests useful, plausible links between gut microbiota and host molecular lipids, the analysis of the fecal metabolome alone is incapable of distinguishing such origins. Bar et al. provides machine-learning algorithms to link factors such as human genetics, diet and microbiome with circulating small molecules in serum [6]. A similar approach may shed light on the factors that affect the origin of microbial small molecules in the gut, including the origin of microbial lipids. Here, the challenge would be to obtain large-scale measurements of several, potentially confounded variables (diet, microbiome, metabolome and other clinical variables), as well as the use of analytical methods for the capture of interactions (gene‑metabolite link) between variables [32]. Besides that, mechanistic studies are required to truly validate these associations [33]. In Section 3, we highlight several studies that mechanistically demonstrate gut microbiota-dependent lipid biotransformation, in particular showing the role of stool microbial species in the regulation of host cholesterol and sphingolipid homeostasis.
3. Synthesis of Lipids by Gut Microbes
Homeostasis between the molecular lipids and gut microbiota is vital for the host metabolic state. Despite a wide range of metabolite classes being produced by the gut microbiota [34[34][35],35], only a few metabolites have lipophilic characteristics, that is, they are able to pass through the epithelial barrier and thus directly impact the host metabolism. Gut microbiota composition and its derived lipids can impact the host metabolic state by altering plasma lipid levels. Here, we discuss specific classes of lipids of microbial or potentially-microbial origin, which are involved in the metabolic interplay between the gut microbiota and the host (Table 1).
Table 1. Significant associations of bioactive lipids, gut microbiome related classes and lipid classes.
| Lipid Category | Lipid Sub Class | Example/Related Lipids | Microbes | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sphingolipids | Ceramide phosphoinositols | PI-Cer(d18:1/22:0) | Bacteroidetes | ( | genera Bacteroides | , | Prevotella | , | Porphyromonas | , | Bacteroides theta | , | thetaiotaomicron | , | ovatus and fragilis | |||||
| Ceramide phosphoethanolamines | N-Acyl ceramide phosphoethanolamine | |||||||||||||||||||
| Sphinganines | 3-ketosphinganine sphinganine |
|||||||||||||||||||
| N | -acylsphinganines | dihydroceramide | ||||||||||||||||||
| C15-, C15OH-, C16OH-, C17OH-, C18:2-, C22:2- dihydrocermide | ||||||||||||||||||||
| Sphingoid base 1-phosphates | sphinganine-1-phosphate (d17:0) sphinganine-1-phosphate (d18:0) |
|||||||||||||||||||
| Sterol | C24 bile acids, alcohols, and derivatives | deoxycholic acid lithocholic acid ursodeoxycholate iso-deoxycholic acid iso-lithocholic acid 7-oxo-lithocholic Acid |
Bacteroides | , | Clostridium | , | Lactobacillus | , and | Bifidobacteria | and | Alloscardovia | sp. | ||||||||
| Taurine conjugates | tauroursodeoxycholic acid | |||||||||||||||||||
| Cholesterol and derivatives | cholestenone coprostanone coprostanol |
Eubacterium coprostanoligenes | , | Bacteroides intestinalis | , | Faecalibacterium prausnitzii | ||||||||||||||
| Fatty Acyls | Other Octadecanoids | 10-hydroxy-12 (Z)-octadecenoic acid (18:1) (HYA), 10-hydroxy-12,15(Z,Z) octadecenoic acid (18:2) (αHYA), 10-hydroxy-6,12(Z,Z)-octadecadienoic acid (18:2) (γHYA), 10-hydroxyoctadecanoic acid (HYB), 10-hydoroxy-trans-11-octade-cenoic acid (HYC), 10-oxo-12(Z)-octadecenoic acid (18:1) (KetoA), 10-oxo-12,15(Z,Z) (18:2) octadecenoic acid (αKetoA), 10-oxo-6,12(Z,Z)-octadecenoic acid (18:2) (γKetoA), 10-oxo-octadecanoic acid, 10-oxo-trans-11-octadecenoic acid |
Lactobacillus genus | ( | Lactobacillus plantarum | , | Lactobacillus salivarius | , | Lactobacillus gasseri | , | Lactobacillus acidophilus | and | Lactobacillus johnsonii | ) | Bifidobacterium | spp., | Eubacterium ventriosum | and | Lactobacillus | spp. |
| Unsaturated fatty acids | oleic acid | |||||||||||||||||||
| Glycerolipids (Endocannabinoid) | Monoacylglycerols | 2-arachidonoylglycerol (2-AG) 2- oleoylglycerol (2-OG) 2- palmitoyl-glycerol (2-PG) |
Akkermansia muciniphila |
3.1. Sphingolipids
SPs are bioactive lipids that regulate various cellular processes including cell differentiation, proliferation, apoptosis and inflammation [36,37][36][37]. In humans, some SPs can be obtained from the diet, while other species are generated by de novo synthesis. The biosynthesis of SPs has been extensively studied in eukaryotes [38]; however, recently the commensal gut microbes (Bacteroides, Prevotella and Porphyromonas) have been reported to produce SPs. Brown et al. reported these microbial derived SPs included ceramide phosphoinositol and deoxy-sphingolipids [33]. Intriguingly, these SPs are reported to aggravate intestinal inflammation and regulate host ceramide pools in animals. In addition, the authors found that microbially-derived SP deficiency was associated with inflammatory bowel disease (IBD), and impacted host-derived SP abundances in humans [33]. Similarly, Johnson et al. mapped the fate of dietary SPs in the gut microbiome, and showed that Bacteroides-derived SPs have an adverse effect on host SP metabolism, specifically on hepatic ceramide levels [39]. Intriguingly, using cell culture, they showed that microbially-derived SPs are incorporated into mammalian SP pathways. More recently, Lee et al. charted the pathways of dietary SPs in the gut microbiome (Figure 1) [40]. Taking the Bioorthogonal labeling-Sort-Seq-Spec (BOSSS) approach, the authors found that dietary SPs were mainly consumed by Bacteroides. They also found that Bifidobacterium, which do not produce SPs, could process dietary SPs in a manner similar to that of SP-producer Bacteroides, suggesting that bioactive lipids which are metabolically-accessible to the gut microbiome could be the target for the host to control its gut microbial composition [40].
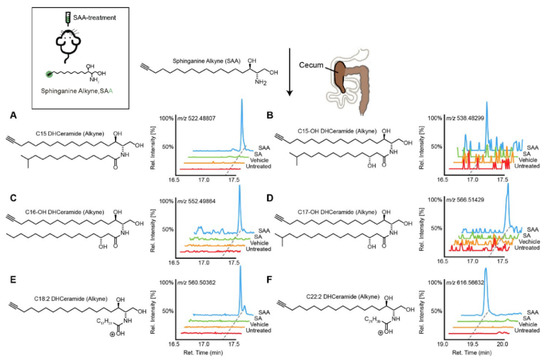
Figure 1. Transformation of dietary sphinganine into dihydrocermides by the gut microbes. Here, sphinganine alkyne (SAA) was given to mice by oral gavage (five consecutive days), then the fecal content from mice was collected and metabolic consequences of SAA exposure were determined using high-resolution mass spectrometry ion chromatograms. The authors showed a distinct cecal lipidome chromatograms for mice that orally treated with SAA (blue), which contained the alkyne-bearing (A) C15-, (B) C15OH-, (C) C16OH-, (D) C17OH-, (E) C18:2- and (F) C22:2- dihydrocermides. However, these dihydrocermides were absent in treatments with sphinganine (SA, green), vehicle or no treatment (red). Figure adapted from [40], with permission under CC BY 4.0 license.
3.2. Sterol
3.2.1. Bile Acids and Derivatives
Primary BAs are produced in hepatocytes; however, secondary BAs such as deoxycholic acid (DCA) and lithocholic acids (LCA), ursodeoxycholate (UDCA) and numerous (≥50) others [41,42][41][42] are produced or biotransformed by gut microbiota. Briefly, in the distal ileum, the conjugated primary BAs are deconjugated by microbial bile salt hydrolases expressed predominantly by anaerobic intestinal bacteria of the genera Bacteroides, Clostridium, Lactobacillus, and Bifidobacteria. This is then followed by 7α-dehydroxylation by a bacterial 7α-dehydroxylase mainly expressed by Clostridium and Eubacterium. Further modifications include oxidation and epimerization of the hydroxyl groups by Bacteroides, Clostridium, Escherichia, Eggerthella, Eubacterium and Peptostreptococcus [43]. Under normal physiological conditions, secondary BA synthesis represents less than 10% of total BA synthesis. Similarly to primary BAs, secondary BAs also modulate host metabolism, the innate immune system in addition to acting as signaling molecules [35,44][35][44]. Ridlon et al. extensively reviewed the emerging BA-gut-microbiome axis [45]. A summary of the key BAs and their microbial linkage are presented in Table 1.
3.2.2. Cholesterol
Cholesterol is a key precursor molecule in the synthesis of many different lipids, including BAs and Vitamin D (fat-soluble secosteroids) [63][46]. Circulating cholesterol is either derived from the diet or produced by de novo synthesis in hepatocytes. Since cholesterol from both sources pass through the intestine, it has been suggested that the gut microbiota modulates plasma cholesterol levels [64,65][47][48]. Recently, by integrating metabolomics and shotgun metagenomics data, Kenny et al. identified the gut microbial enzymes involved in cholesterol metabolism (Figure 2) [52][49]. They found that the cholesterol dehydrogenases enzyme (encoded by ismA genes of the gut microbiota) has a significant impact on both fecal and total serum cholesterol levels in humans [52][49]. However, whether these metabolically-active gut microbes are linked to the diet or not remains unknown. In the Dutch LifeLines-DEEP cohort involving 893 human subjects, Fu et al. showed that the gut microbiome contributed a substantial proportion of the variation in circulating high-density lipoprotein cholesterol (HDL), but not to total cholesterol or low-density lipoprotein (LDL) cholesterol levels [9]. Taken together, these studies suggest that cholesterol metabolism by gut microbes is crucial in host cholesterol homeostasis (Figure 2) [52][49].
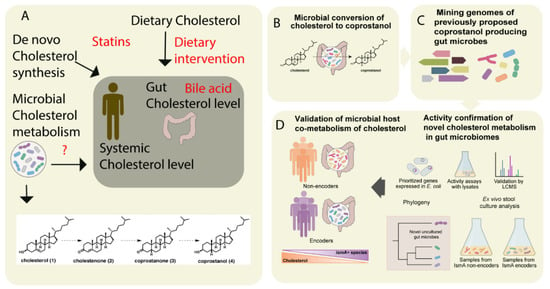
Figure 2. The metabolism of cholesterol by gut microbes influences both intestinal and circulating cholesterol concentrations. (A) The cholesterol level in the blood can be influenced either by de novo cholesterol synthesized in the liver or it may be derived from exogenous sources such as the diet. The endogenous cholesterol level may be also influenced by drugs such as statins, or via altered bile acid metabolism. In addition, gut microbial metabolism of cholesterol may also serve as check point for the maintenance of cholesterol homeostasis (B). As shown in panel A, the authors proposed a pathway for microbial conversion of cholesterol (1) to coprostanol (4) in the microbiota involves the intermediates cholestenone (2) and coprostanone (3). (C) By using genome mining, Kenny et al. identified and characterized microbial enzymes linked with cholesterol metabolism. Later, integrating the functional metagenomics and metabolomics data, they predicted as well as validated a group of microbes and their microbial activities (microbial cholesterol dehydrogenases) which mediated the metabolism of cholesterol in the gut (D). Figure adapted from [52][49] with permission under CC BY 4.0 license.
3.3. Fatty Acyls and Conjugates
Fatty acids produced by gut microbiota can stimulate synthesis of mono-unsaturated fatty acids (MUFAs) and the elongation of polyunsaturated fatty acids (PUFAs) in the host. A multi-omics profiling study found that circulating lipid levels can be affected by microbial fatty acid metabolism [66][50]. MUFA (16:1n−7, FA 18:1n−9, 18:1n−7), MUPC (PC 34:1, PC 36:1) and PUFA (20:3n−6, 22:6−3) levels were associated with acetate production in the gut. In addition to their free forms, fatty acids can also be found conjugated to mono-amine neuro-transmitters, such as serotonin, forming arachidonoyl-serotonin (AA-5-HT), oleoyl-serotonin, palmitoyl-serotonin, and stearoyl-serotonin. The neurotransmitter serotonin (5-hydroxytryptamine) in humans is mainly (90%) synthesized in the gastrointestinal tract by human gut microbiota [67][51] (see also related Section 3.4 below, on endocannabinoids). Serotonin and SCFA concentrations stimulate the formation of N-acyl ethanolamine (NAE) conjugates [68][52], which form a novel class of endogenous lipid mediators in the intestine. Emerging evidence also indicates a role for the intestinal microbiota in the production of N-acyl serotonin’s metabolites, which have been shown to modulate the enteric nervous system [69,70][53][54]. Additionally, conjugated fatty acids are also formed by commensal gut microbes such as Lactobacillus plantarum [53][55]. In particular, conjugated linoleic acids (CLAs), such as conjugated diene structures cis-9, trans-11-CLA and trans-9,trans-11-CLA, are formed in the gastrointestinal tract passing through the intermediate 10-hydroxy-12-octadecenoic acid [53][55].
3.4. Endocannabinoids
The endocannabinoid system is comprised of lipid-derived endogenous cannabinoid receptor ligands (endocannabinoids), enzymes involved in their synthesis and degradation, and the cannabinoid 1 and 2 receptors (CB1R and CB2R), which have differential affinities for endocannabinoids. Growing evidence suggests that gut microbes and the endocannabinoid system are interlinked [58,71][56][57]. Rousseaux et al. initially proposed the link between the gut microbes (Lactobacillus acidophilus) and the endocannabinoid system [71][57]. Later, Cani et al. demonstrated indirect crosstalk between the gut microbiota and the endocannabinoid system, which modulated host adipogenesis [72][58]. The authors found that peripheral (e.g., intestine and adipose tissue) endocannabinoid (specifically, anandamide; AEA) levels were influenced by gut microbiota in the experimental animals. In another study, Everard et al. showed that A. muciniphila (which represents 3–5% of the human microbial community) treatment in obese mice increased the levels of glycerolipids intestinal 2- oleoylglycerol (2-OG), 2-arachidonoylglycerol (2-AG) and 2- palmitoyl-glycerol (2-PG) [73][59]. More recently, endocannabinoid–like molecule N-acyl-3-hydroxypalmitoyl-glycine (commendamide) was reported to be produced by the commensal microbe Bacteroides [74,75][60][61]. These findings suggest that specific gut microbes produce specific classes of lipids which impact these host signaling and metabolic pathways.
3.5. Carnitine and Acyl Carnitines
De novo synthesis of carnitines occurs in all domains of life [76][62]. Carnitines that are not digested or absorbed in the small intestine are excreted to the large intestine, where they are catabolized by gut microbes. Some microbes (Pseudomonas species) utilize carnitine as a source of carbon and nitrogen, while Acinetobacter calcoaceticus catabolize carnitine into trimethylamine (TMA), which is then converted into trimethylamine N-oxide (TMAO) by hepatic flavin monooxygenases (FMOs). TMAO is strongly linked with cardiovascular diseases [77,78][63][64]. Also, phosphatidylcholines (PCs), a major a class of phospholipids, forming the major structural components of cellular plasma membranes are also converted to trimethylamine (TMA) by gut microbes. These PCs are abundant in foods such as fish, eggs and milk, suggesting that dietary lipids regulate host lipid metabolism through interaction with the gut microbiota [79][65]. Interestingly, Hulme et al. identified two structural analogs of carnitine, 3-methyl-4-(trimethylammonio) butanoate and 4-(trimethylammonio) pentanoate, which are produced by anaerobic commensals from the Clostridiales family [80][66]. Acyl carnitines (fatty acyl esters of L-carnitine) have also been reported in human feces [81,82][67][68]. However, information about the microbes that facilitate the biotransformation of acyl carnitines in the gut is still unknown [52,58,80][49][56][66].
References
- Tran, T.T.; Postal, B.G.; Demignot, S.; Ribeiro, A.; Osinski, C.; Pais de Barros, J.P.; Blachnio-Zabielska, A.; Leturque, A.; Rousset, M.; Ferre, P.; et al. Short Term Palmitate Supply Impairs Intestinal Insulin Signaling via Ceramide Production. J. Biol. Chem. 2016, 291, 16328–16338.
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194.
- Wei, F.; Lamichhane, S.; Orešič, M.; Hyötyläinen, T. Lipidomes in health and disease: Analytical strategies and considerations. TrAC Trends Anal. Chem. 2019, 120, 115664.
- VanHook, A.M. Microbial metabolites shape lipid metabolism. Sci. Signal. 2020, 13, eabc1552.
- Visconti, A.; Le Roy, C.I.; Rosa, F.; Rossi, N.; Martin, T.C.; Mohney, R.P.; Li, W.; de Rinaldis, E.; Bell, J.T.; Venter, J.C.; et al. Interplay between the human gut microbiome and host metabolism. Nat. Commun. 2019, 10, 4505.
- Bar, N.; Korem, T.; Weissbrod, O.; Zeevi, D.; Rothschild, D.; Leviatan, S.; Kosower, N.; Lotan-Pompan, M.; Weinberger, A.; Le Roy, C.I.; et al. A reference map of potential determinants for the human serum metabolome. Nature 2020.
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.-M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273.
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381.
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 2015, 117, 817–824.
- Liu, H.; Pan, L.L.; Lv, S.; Yang, Q.; Zhang, H.; Chen, W.; Lv, Z.; Sun, J. Alterations of Gut Microbiota and Blood Lipidome in Gestational Diabetes Mellitus With Hyperlipidemia. Front. Physiol. 2019, 10, 1015.
- Benítez-Páez, A.; Kjølbæk, L.; Gómez Del Pulgar, E.M.; Brahe, L.K.; Astrup, A.; Matysik, S.; Schött, H.F.; Krautbauer, S.; Liebisch, G.; Boberska, J.; et al. A Multi-omics Approach to Unraveling the Microbiome-Mediated Effects of Arabinoxylan Oligosaccharides in Overweight Humans. mSystems 2019, 4.
- Albouery, M.; Buteau, B.; Gregoire, S.; Cherbuy, C.; Pais de Barros, J.P.; Martine, L.; Chain, F.; Cabaret, S.; Berdeaux, O.; Bron, A.M.; et al. Age-Related Changes in the Gut Microbiota Modify Brain Lipid Composition. Front. Cell. Infect. Microbiol. 2019, 9, 444.
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R.; et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134.
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Orešič, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018, 149, 3–12.
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24.
- Gregory, K.E.; Bird, S.S.; Gross, V.S.; Marur, V.R.; Lazarev, A.V.; Walker, W.A.; Kristal, B.S. Method development for fecal lipidomics profiling. Anal. Chem. 2013, 85, 1114–1123.
- Van Meulebroek, L.; De Paepe, E.; Vercruysse, V.; Pomian, B.; Bos, S.; Lapauw, B.; Vanhaecke, L. Holistic Lipidomics of the Human Gut Phenotype Using Validated Ultra-High-Performance Liquid Chromatography Coupled to Hybrid Orbitrap Mass Spectrometry. Anal. Chem. 2017, 89, 12502–12510.
- Trost, K.; Ahonen, L.; Suvitaival, T.; Christiansen, N.; Nielsen, T.; Thiele, M.; Jacobsen, S.; Krag, A.; Rossing, P.; Hansen, T.; et al. Describing the fecal metabolome in cryogenically collected samples from healthy participants. Sci. Rep. 2020, 10, 885.
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288.
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Hyötyläinen, T.; Orešič, M. An overview of metabolomics data analysis: Current tools and future perspectives. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 82, pp. 387–2413.
- Zullig, T.; Trotzmuller, M.; Kofeler, H.C. Lipidomics from sample preparation to data analysis: A primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209.
- O’Shea, K.; Misra, B.B. Software tools, databases and resources in metabolomics: Updates from 2018 to 2019. Metabolomics 2020, 16, 36.
- Gao, X.; Pujos-Guillot, E.; Sebedio, J.L. Development of a quantitative metabolomic approach to study clinical human fecal water metabolome based on trimethylsilylation derivatization and GC/MS analysis. Anal. Chem. 2010, 82, 6447–6456.
- Phua, L.C.; Koh, P.K.; Cheah, P.Y.; Ho, H.K.; Chan, E.C. Global gas chromatography/time-of-flight mass spectrometry (GC/TOFMS)-based metabonomic profiling of lyophilized human feces. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 937, 103–113.
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048.
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dormann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. : The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638.
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617.
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612.
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164.
- Gil-de-la-Fuente, A.; Godzien, J.; Saugar, S.; Garcia-Carmona, R.; Badran, H.; Wishart, D.S.; Barbas, C.; Otero, A. CEU Mass Mediator 3.0: A Metabolite Annotation Tool. J. Proteome Res. 2019, 18, 797–802.
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795.
- Bradley, P.H.; Pollard, K.S. Building a chemical blueprint for human blood. Nature 2020, 588, 36–37.
- Brown, E.M.; Ke, X.; Hitchcock, D.; Jeanfavre, S.; Avila-Pacheco, J.; Nakata, T.; Arthur, T.D.; Fornelos, N.; Heim, C.; Franzosa, E.A.; et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 2019, 25, 668–680.
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91.
- Cani, P.D.; Van Hul, M.; Lefort, C.; Depommier, C.; Rastelli, M.; Everard, A. Microbial regulation of organismal energy homeostasis. Nat. Metab. 2019, 1, 34–46.
- Hannun, Y.A.; Obeid, L.M. Author Correction: Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 673.
- Kolter, T. A view on sphingolipids and disease. Chem. Phys. Lipids 2011, 164, 590–606.
- Heaver, S.L.; Johnson, E.L.; Ley, R.E. Sphingolipids in host-microbial interactions. Curr. Opin. Microbiol. 2018, 43, 92–99.
- Johnson, E.L.; Heaver, S.L.; Waters, J.L.; Kim, B.I.; Bretin, A.; Goodman, A.L.; Gewirtz, A.T.; Worgall, T.S.; Ley, R.E. Sphingolipids produced by gut bacteria enter host metabolic pathways impacting ceramide levels. Nat. Commun. 2020, 11, 2471.
- Lee, M.T.; Le, H.H.; Johnson, E.L. Dietary sphinganine is selectively assimilated by members of the mammalian gut microbiome. J. Lipid Res. 2020.
- Devlin, A.S.; Fischbach, M.A. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 2015, 11, 685–690.
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259.
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853.
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The Role of the Gut Microbiota in Bile Acid Metabolism. Ann. Hepatol. 2017, 16, s15–s20.
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338.
- Yang, S.T.; Kreutzberger, A.J.B.; Lee, J.; Kiessling, V.; Tamm, L.K. The role of cholesterol in membrane fusion. Chem. Phys. Lipids 2016, 199, 136–143.
- Kriaa, A.; Bourgin, M.; Potiron, A.; Mkaouar, H.; Jablaoui, A.; Gérard, P.; Maguin, E.; Rhimi, M. Microbial impact on cholesterol and bile acid metabolism: Current status and future prospects. J. Lipid Res. 2019, 60, 323–332.
- Koppel, N.; Maini Rekdal, V.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356.
- Kenny, D.J.; Plichta, D.R.; Shungin, D.; Koppel, N.; Hall, A.B.; Fu, B.; Vasan, R.S.; Shaw, S.Y.; Vlamakis, H.; Balskus, E.P.; et al. Cholesterol Metabolism by Uncultured Human Gut Bacteria Influences Host Cholesterol Level. Cell Host Microbe 2020, 28, 245–257.
- Kindt, A.; Liebisch, G.; Clavel, T.; Haller, D.; Hörmannsperger, G.; Yoon, H.; Kolmeder, D.; Sigruener, A.; Krautbauer, S.; Seeliger, C.; et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nat. Commun. 2018, 9, 3760.
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403.
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366.
- Verhoeckx, K.C.; Voortman, T.; Balvers, M.G.; Hendriks, H.F.; Wortelboer, H.M.; Witkamp, R.F. Presence, formation and putative biological activities of N-acyl serotonins, a novel class of fatty-acid derived mediators, in the intestinal tract. Biochim. Biophys. Acta 2011, 1811, 578–586.
- Sharon, G.; Garg, N.; Debelius, J.; Knight, R.; Dorrestein, P.C.; Mazmanian, S.K. Specialized metabolites from the microbiome in health and disease. Cell Metab. 2014, 20, 719–730.
- Kishino, S.; Takeuchi, M.; Park, S.B.; Hirata, A.; Kitamura, N.; Kunisawa, J.; Kiyono, H.; Iwamoto, R.; Isobe, Y.; Arita, M.; et al. Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc. Natl. Acad. Sci. USA 2013, 110, 17808–17813.
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids--at the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143.
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37.
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392.
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071.
- Lynch, A.; Crowley, E.; Casey, E.; Cano, R.; Shanahan, R.; McGlacken, G.; Marchesi, J.R.; Clarke, D.J. The Bacteroidales produce an N-acylated derivative of glycine with both cholesterol-solubilising and hemolytic activity. Sci. Rep. 2017, 7, 13270.
- Cohen, L.J.; Kang, H.S.; Chu, J.; Huang, Y.H.; Gordon, E.A.; Reddy, B.V.; Ternei, M.A.; Craig, J.W.; Brady, S.F. Functional metagenomic discovery of bacterial effectors in the human microbiome and isolation of commendamide, a GPCR G2A/132 agonist. Proc. Natl. Acad. Sci. USA 2015, 112, E4825–E4834.
- Meadows, J.A.; Wargo, M.J. Carnitine in bacterial physiology and metabolism. Microbiology 2015, 161, 1161–1174.
- Ghonimy, A.; Zhang, D.M.; Farouk, M.H.; Wang, Q. The Impact of Carnitine on Dietary Fiber and Gut Bacteria Metabolism and Their Mutual Interaction in Monogastrics. Int. J. Mol. Sci. 2018, 19, 1008.
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154.
- Sitaraman, R. Phospholipid catabolism by gut microbiota and the risk of cardiovascular disease. J. Med. Microbiol. 2013, 62, 948–950.
- Hulme, H.; Meikle, L.M.; Strittmatter, N.; van der Hooft, J.J.J.; Swales, J.; Bragg, R.A.; Villar, V.H.; Ormsby, M.J.; Barnes, S.; Brown, S.L.; et al. Microbiome-derived carnitine mimics as previously unknown mediators of gut-brain axis communication. Sci. Adv. 2020, 6, eaax6328.
- Turroni, S.; Fiori, J.; Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Barone, M.; Biagi, E.; Fanelli, F.; Mezzullo, M.; Crittenden, A.N.; et al. Fecal metabolome of the Hadza hunter-gatherers: A host-microbiome integrative view. Sci. Rep. 2016, 6, 32826.
- Huang, H.J.; Zhang, A.Y.; Cao, H.C.; Lu, H.F.; Wang, B.H.; Xie, Q.; Xu, W.; Li, L.J. Metabolomic analyses of faeces reveals malabsorption in cirrhotic patients. Dig. Liver Dis. 2013, 45, 677–682.