Sequencing of DNA from single isolated chromosomes (ChromSeq) is an elegant approach to determine the chromosome content and assign genome assemblies to chromosomes, thus bridging the gap between cytogenetics and genomics.
- ChromSeq
1. Introduction
Single-chromosome sequencing has been previously referred to as ChromSeq in plant genome studies.
2. ChromSeq Workflow
ChromSeq workflow consists in three main steps: (i) physical chromosome isolation; (ii) high throughput sequencing of isolated chromosomal DNA; and (iii) bioinformatic analysis of sequencing data (Figure 1).
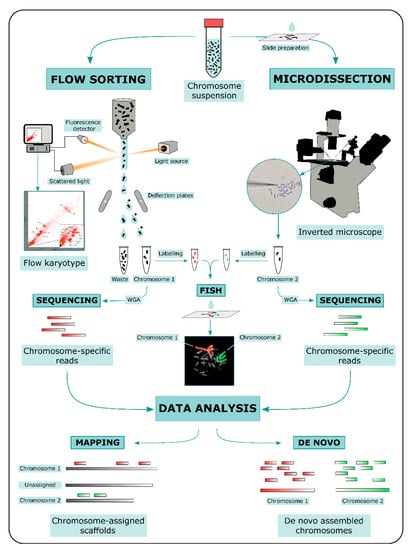
Schematic representation of ChromSeq workflow. Briefly, chromosomes are isolated via either flow sorting or microdissection (only mechanical microdissection is shown). After isolation, Whole-Genome Amplification (WGA) is performed on chromosomal DNA. Eventually, chromosomal DNA can be labeled with fluorochromes and hybridized onto the target species metaphases to confirm the identity of isolated chromosomes. WGA products are then sequenced with next generation sequencing technologies. Sequencing data can be mapped on the target species reference genome or assembled
. The latter approach has proven successful when a combination of high throughput chromosome isolation (millions of copies) and long-read sequencing approaches are implemented.
Two main methods for physical chromosome isolation are currently available: flow sorting and microdissection [38][1]. Both approaches require the preparation of a metaphase chromosome suspension. Other methods based on microfluidic mechanics have been developed in the last decade for chromosome isolation (e.g., [39,40,41][2][3][4]); however, these methods were not widely used compared to flow sorting and microdissection.
In flow sorting, chromosomal DNA is labeled with two different fluorochromes specific for GC- and AT-rich regions. Fluorochrome-labeled chromosomes are passed through a narrow stream of liquid and broken into fine droplets. Fluorescence intensity is measured for each chromosome contained in a droplet, and the measurements of fluorescence intensities are visualized as a flow karyotype. Ideally, each chromosome forms a distinct peak in the flow karyotype, whose location is proportional to the ratio of GC/AT fluorescence intensity, a relative measure of chromosome size. Peaks can be gated for a specific fluorescence intensity ratio and droplets containing single chromosomes are deflected with an electromagnetic field into tubes [32,42,43][5][6][7]. An advantage of this method is the possibility to isolate a high number of specific chromosomes. However, key disadvantages of the method are the difficulty to separate chromosomes with similar size, and impurity due to the fragmentation of chromosomes.
In microdissection, metaphase spreads on slides are used. Metaphases are observed under an inverted microscope and single chromosomes are physically isolated either with a micromanipulator armed with thin glass needles (mechanical microdissection) or cut out with a laser beam (laser microdissection). In laser microdissection slides are covered with specific membranes to allow the chromosome cut [38,44][1][8]. This method generates relatively contamination-free samples and can be used to isolate not only a whole chromosome, but also specific target regions [45,46][9][10]. However, microdissection is labor intensive and is restricted to the isolation of usually no more than a dozen chromosomes per type. Moreover, part of the chromosomal DNA can be damaged or lost during the isolation.
Both flow sorting and microdissection yield DNA quantities, which are by themselves too low for high-throughput sequencing. For this reason Whole-Genome Amplification (WGA) is performed on chromosomal DNA prior to sequencing using either degenerate primer (DOP-PCR, Ref. [47][11]) or multiple displacement amplification (MDA, Refs. [48,49][12][13]). An aliquot of the amplified DNA can be used to produce chromosome paints [50][14]. For this purpose, amplified DNA is labeled with fluorochromes and eventually hybridized onto the target species metaphases, in order to confirm the identity of isolated chromosomes (e.g., [51][15]). Once a clear correspondence between the isolated chromosomal DNA and the species karyotype is obtained through FISH of chromosome paints, amplified DNA can be used to prepare libraries for high-throughput sequencing according to the manufacturer’s protocols. Currently a short-read sequencing approach is mainly preferred for ChromSeq (e.g., [52,53,54,55,56][16][17][18][19][20]), but long-read approaches have also been employed (e.g., [57][21]).
Sequencing data generated from isolated chromosomes can be processed using a wide variety of approaches that can be divided into two main categories: (i) alignment to a reference genome and (ii) de novo assembly of chromosome-specific sequencing data. In cases DOP-PCR or MDA are used prior to the chromosome-specific library sequencing, pre-processing of sequencing data is needed to trim primers and/or adapters independently from the approach used.
Reference-based analysis consists in the alignment of chromosome-specific reads to a reference genome, and represents the most commonly used approach so far. Based on the alignment data, reference genome scaffolds are assigned to specific chromosomes, that is, if reads obtained from chromosome 1 map onto three different scaffolds of the reference genome, it means that those three scaffolds are parts of chromosome 1. If no rearrangement is expected between the reference genome and the sampled chromosome, any statistic for mapped read density can be used to rank scaffolds and subsequently retain those assigned to the chromosome. The problem is further complicated if rearrangements between the target species chromosome and reference genome are possible. In order to predict the rearrangement breakpoints, several methods were successfully developed based on various statistical approaches and read density metrics, including maximum likelihood based on read count per Kb [29[22][23],58], circular binary segmentation [56][20] or clustering [59][24] based on distances between non-overlapping read mappings. The software DOPseq is the only one developed ad hoc for ChromSeq data analysis and it unifies the chromosome region detection with the upstream processing into an automated and reproducible pipeline [56][20]. The main disadvantages of a reference-based approach involve errors in read mapping and sample contamination, which can lead to misinterpretation of alignment data. Therefore, it is crucial to separate the true chromosome assignment signal from background noise.
A de novo assembly approach can also be implemented on ChromSeq data. In this case, chromosome-specific assemblies are produced independently for each chromosome. This approach requires cross-contamination checks among all chromosome-specific data pools and repetitive sequence removal to increase the assembly contiguity (e.g., [60][25]). Sequencing data derived from only a few isolated chromosomes are usually highly fragmented and a de novo approach might produce assembly with a low contig N50 (e.g., [61][26]). However, this problem can be circumvented by either sequencing a very large number of chromosome copies (up to millions) or by implementing sequencing data with long-read approaches. Kuderna et al. [62][27] for instance, successfully assembled de novo the human chromosome 1 by using a combination of high throughput chromosome isolation (10 million copies) and Oxford Nanopore sequencing. The resulting assembly had an N50 of 10.5 Mb and allowed the identification of structural variants. The gorilla Y chromosome was also successfully assembled de novo using a combination of short and long-read sequencing [57][21].
References
- Yang, F.; Trifonov, V.; Ng, B.L.; Kosyakova, N.; Carter, N.P. Generation of paint probes from flow-sorted and microdissected chromosomes. In Fluorescence In Situ Hybridization (FISH); Liehr, T., Ed.; Springer: Berlin, Germany, 2017; pp. 63–79.
- Benítez, J.J.; Topolancik, J.; Tian, H.C.; Wallin, C.B.; Latulippe, D.R.; Szeto, K.; Murphy, P.J.; Cipriany, B.R.; Levy, S.L.; Soloway, P.D.; et al. Microfluidic extraction, stretching and analysis of human chromosomal DNA from single cells. Lab Chip 2012, 12, 4848–4854.
- Fan, H.C.; Wang, J.; Potanina, A.; Quake, S.R. Whole-genome molecular haplotyping of single cells. Nat. Biotechnol. 2011, 29, 51–57.
- Takahashi, T.; Okeyo, K.O.; Ueda, J.; Yamagata, K.; Washizu, M.; Oana, H. A microfluidic device for isolating intact chromosomes from single mammalian cells and probing their folding stability by controlling solution conditions. Sci. Rep. 2018, 8, 13684.
- Doležel, J.; Vrána, J.; Šafář, J.; Bartoš, J.; Kubaláková, M.; Šimková, H. Chromosomes in the flow to simplify genome analysis. Funct. Integr. Genom. 2012, 12, 397–416.
- Ibrahim, S.F.; van den Engh, G. High-speed chromosome sorting. Chrom. Res. 2004, 12, 5–14.
- Stanyon, R.; Stone, G. Phylogenomic analysis by chromosome sorting and painting. In Phylogenomics. Methods in Molecular Biology; William, J.M., Ed.; Springer: Berlin, Germany, 2008; Volume 422, pp. 13–29.
- Zhou, R.-N.; Hu, Z.-M. The development of chromosome microdissection and microcloning technique and its applications in genomic research. Curr. Genom. 2007, 8, 67–72.
- Weise, A.; Timmermann, B.; Grabherr, M.; Werber, M.; Heyn, P.; Kosyakova, N.; Liehr, T.; Neitzel, H.; Konrat, K.; Bommer, C.; et al. High-throughput sequencing of microdissected chromosomal regions. Eur. J. Hum. Genet. 2010, 18, 457–462.
- Zlotina, A.; Maslova, A.; Pavlova, O.; Kosyakova, N.; Al-Rikabi, A.; Liehr, T.; Krasikova, A. New Insights Into Chromomere Organization Provided by Lampbrush Chromosome Microdissection and High-Throughput Sequencing. Front. Genet. 2020, 11, 57.
- Telenius, H.; Ponder, B.A.J.; Tunnacliffe, A.; Pelmear, A.H.; Carter, N.P.; Ferguson-Smith, M.A.; Behmel, A.; Nordenskjöld, M.; Pfragner, R. Cytogenetic analysis by chromosome painting using DOP-PCR amplified flow-sorted chromosomes. Genes Chromosom. Cancer 1992, 4, 257–263.
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266.
- Zhang, K.; Martiny, A.C.; Reppas, N.B.; Barry, K.W.; Malek, J.; Chisholm, S.W.; Church, G.M. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006, 24, 680–686.
- Ried, T.; Schröck, E.; Ning, Y.; Wienberg, J. Chromosome painting: A useful art. Hum. Mol. Genet. 1998, 7, 1619–1626.
- Teer, J.K.; Johnston, J.J.; Anzick, S.L.; Pineda, M.; Stone, G.; Meltzer, P.S.; Mullikin, J.C.; Biesecker, L.G. Massively-parallel sequencing of genes on a single chromosome: A comparison of solution hybrid selection and flow sorting. BMC Genom. 2013, 14, 1–10.
- Andreyushkova, D.A.; Makunin, A.I.; Beklemisheva, V.R.; Romanenko, S.A.; Druzhkova, A.S.; Biltueva, L.B.; Serdyukova, N.A.; Graphodatsky, A.S.; Trifonov, V.A. Next generation sequencing of chromosome-specific libraries sheds light on genome evolution in paleotetraploid sterlet (Acipenser ruthenus). Genes 2017, 8, 318.
- Kichigin, I.G.; Giovannotti, M.; Makunin, A.I.; Ng, B.L.; Kabilov, M.R.; Tupikin, A.E.; Barucchi, V.C.; Splendiani, A.; Ruggeri, P.; Rens, W.; et al. Evolutionary dynamics of Anolis sex chromosomes revealed by sequencing of flow sorting-derived microchromosome-specific DNA. Mol. Genet. Genom. 2016, 291, 1955–1966.
- Lind, A.L.; Lai, Y.Y.Y.; Mostovoy, Y.; Holloway, A.K.; Iannucci, A.; Mak, A.C.Y.; Fondi, M.; Orlandini, V.; Eckalbar, W.L.; Milan, M.; et al. Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 2019, 3, 1241–1252.
- Lisachov, A.P.; Makunin, A.I.; Giovannotti, M.; Pereira, J.C.; Druzhkova, A.S.; Barucchi, V.C.; Ferguson-Smith, M.A.; Trifonov, V.A. Genetic content of the neo-sex chromosomes in Ctenonotus and Norops (Squamata, Dactyloidae) and degeneration of the Y chromosome as revealed by high-throughput sequencing of individual chromosomes. Cytogenet. Genome Res. 2019, 157, 115–122.
- Makunin, A.I.; Kichigin, I.G.; Larkin, D.M.; O’Brien, P.C.; Ferguson-Smith, M.A.; Yang, F.; Proskuryakova, A.A.; Vorobieva, N.V.; Chernyaeva, E.N.; O’Brien, S.J.; et al. Contrasting origin of B chromosomes in two cervids (Siberian roe deer and grey brocket deer) unravelled by chromosome-specific DNA sequencing. BMC Genom. 2016, 17, 618.
- Tomaszkiewicz, M.; Rangavittal, S.; Cechova, M.; Campos Sanchez, R.; Fescemyer, H.W.; Harris, R.; Ye, D.; O’Brien, P.C.M.; Chikhi, R.; Ryder, O.A.; et al. A time-and cost-effective strategy to sequence mammalian Y Chromosomes: An application to the de novo assembly of gorilla Y. Genome Res. 2016, 26, 530–540.
- Chen, W.; Kalscheuer, V.; Tzschach, A.; Menzel, C.; Ullmann, R.; Schulz, M.H.; Erdogan, F.; Li, N.; Kijas, Z.; Arkesteijn, G.; et al. Mapping translocation breakpoints by next-generation sequencing. Genome Res. 2008, 18, 1143–1149.
- Chen, W.; Ullmann, R.; Langnick, C.; Menzel, C.; Wotschofsky, Z.; Hu, H.; Döring, A.; Hu, Y.; Kang, H.; Tzschach, A.; et al. Breakpoint analysis of balanced chromosome rearrangements by next-generation paired-end sequencing. Eur. J. Hum. Genet. 2010, 18, 539–543.
- Komissarov, A.; Vij, S.; Yurchenko, A.; Trifonov, V.; Thevasagayam, N.; Saju, J.; Sridatta, P.S.R.; Purushothaman, K.; Graphodatsky, A.; Orbán, L.; et al. B Chromosomes of the Asian seabass (Lates calcarifer) contribute to genome variations at the level of individuals and populations. Genes 2018, 9, 464.
- Brinkrolf, K.; Rupp, O.; Laux, H.; Kollin, F.; Ernst, W.; Linke, B.; Kofler, R.; Romand, S.; Hesse, F.; Budach, W.E.; et al. Chinese hamster genome sequenced from sorted chromosomes. Nat. Biotechnol. 2013, 31, 694–695.
- Luo, W.; Xia, Y.; Yue, B.; Zeng, X. Assigning the sex-specific markers via genotyping-by-sequencing onto the Y chromosome for a torrent frog Amolops mantzorum. Genes 2020, 11, 727.
- Kuderna, L.F.K.; Solís-Moruno, M.; Batlle-Masó, L.; Julià, E.; Lizano, E.; Anglada, R.; Ramírez, E.; Bote, A.; Tormo, M.; Marquès-Bonet, T.; et al. Flow sorting enrichment and nanopore sequencing of chromosome 1 from a Chinese individual. Front. Genet. 2020, 10, 1315.