The complement system is an essential component of innate immunity. Acting as a first line of defence, the complement system recognises and aids in the elimination of pathogens. Complement proteins also play key roles in homeostasis and they support the inductive of the adaptive immune response.
- complement
- cancer
- cancer treatment
- therapeutic response
1. Introduction
A dynamic relationship exists between the immune system and cancer, owing to the fact that a system designed to defend the host and maintain homeostasis has the potential to promote and foster malignant transformation [1][2][1,2]. Complement, an innate inflammatory system, is no exception to this paradox [3]. Traditionally, the elimination of foreign antigens was considered the primary, if not sole function of complement, however we now understand that complement activities extend beyond this [4]. The complement system, for instance, plays an important role coordinating adaptive immune responses, as an opsonin, in synapse elimination and during angiogenesis [5][6][7][8][5,6,7,8]. Several studies have demonstrated that complement is also capable of recognising and eliminating malignant cells [9]. The net effect of these diverse functions renders the complement system a key player in immune surveillance and homeostasis [4]. The delicate equilibrium between developing tumours and the immune system is well documented, with evasion of immune destruction defined as a hallmark of cancer [10]. In line with reports of an altered immune milieu in several human cancers, dysregulation of the complement system in the cancer setting has been observed [11][12][13][14][15][11,12,13,14,15]. More recently, pro-oncogenic roles for complement cascade components have been described [16][17][16,17]. Analysis of the current literature suggests that the complement system has a dual role in malignancy [4][18][4,18] and whether complement protects against or enables tumour pathogenesis may depend on the context of the tumour microenvironment (TME) [19].
2. The Complement System
In 1901, Jules Bordet described complement as a heat-labile factor that augmented antibody-mediated bacterial lysis [20]. Subsequent discoveries have since established that complement is not a single entity but represents a family of many proteins [21]. The complement system is composed of approximately 50 soluble and membrane-bound complement effectors, regulators and receptors, with the main complement proteins numbered C1-9 [4]. Many complement precursors exist as zymogens, which require cleavage in order to gain functionality [22]. The C3 and C5 convertase enzymes are central to the complement cascade, cleaving C3 and C5 respectively to generate anaphylatoxins (C3a, C5a) and opsonins (C3b, C5b) [4][23][4,23]. The small anaphylatoxin molecules are potent inflammatory mediators with many effector functions [22][24][22,24]. Complement proteins are primarily produced by the liver before systemic dissemination via the bloodstream, however, we now understand that T cells, macrophages, endothelial cells and more recently, cancer cells, are capable of complement production.
2.1. Complement Activation Pathways
There are three pathways by which the complement system may be activated, the classical, the lectin and the alternative pathways (Figure 1). The classical pathway is principally initiated when C1q of the C1 complex (C1q, C1r and C1s) recognises antigen-antibody (Immunoglobulin (Ig) G or IgM containing) immune complexes, but several antibody-independent signals such as C-reactive protein and viral proteins can activate this pathway also [25][26][27][28][29][30][31][25,26,27,28,29,30,31]. Viral and bacterial carbohydrate-based pathogen-associated molecular patterns (PAMPs) activate the lectin pathway by binding to mannose-binding lectin (MBL), ficolins or collectins [32][33][34][35][32,33,34,35]. In the alternative pathway, C3 is spontaneously hydrolysed to C3H2O in a process known as ‘tick-over’ [36][37][36,37]. Bacterial and yeast polysaccharides and damaged tissue are among the initiators of this pathway [38][39][38,39]. The binding of properdin to target microbial surfaces can facilitate the assembly of the alternative pathway C3 convertase [40][41][40,41]. The alternative pathway also acts as an amplification loop for the classical and lectin pathways [23][42][23,42]. Irrespective of the course of activation, the three activation pathways converge to initiate the terminal pathway. This pathway culminates with the assembly of complement components C5b–C9 to form a membrane attack complex (MAC) [43][44][43,44]. MAC insertion into target cell membranes can trigger lysis known as complement-dependent cytotoxicity (CDC) [45], or at sublytic doses may activate signalling pathways to promote cell survival [46][47][46,47].
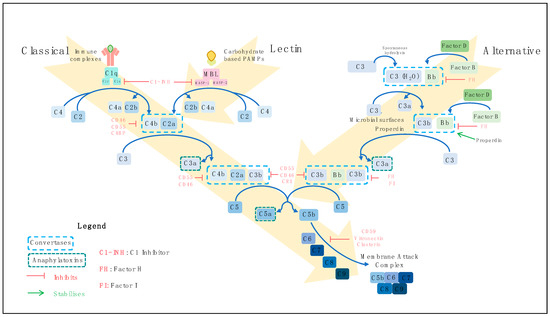
Figure 1. Complement activation pathways. There are three routes by which the complement system can become activated: the classical, the lectin and the alternative pathways. Classical pathway activation is initiated primarily by antigen-antibody immune complexes. C1q of the C1 complex (C1q, C1r and C1s) interacts with the fragment crystallistion (Fc) portion of antigen-bound immunoglobulins, activating C1r, which subsequently cleaves and activates C1s. Activated C1s cleaves C4 into C4a and C4b, and C2 into C2a and C2b leading to assembly of C4bC2a, the C3 convertase. Carbohydrate-based pathogen-associated molecular patterns (PAMPs) trigger activation of the lectin pathway. Mannose-binding lectin (MBL), ficolins or collectins recognise PAMPs, activating MBL-associated serine proteases (MASPs). Similar to the classical pathway, C4 and C2 are cleaved to generate C4bC2a. The classical and lectin complement activation pathways converge at this point to cleave C3 into the potent anaphylatoxin C3a, and C3b, which joins the C3 convertase to form C4bC2aC3b, the C5 convertase. Cleavage of C5 yields the C5a anaphylatoxin and C5b, which polymerises with C6, C7, C8 and C9 to form the membrane-attack complex (MAC). This inserts into target cell membranes to induce lysis. Spontaneous hydrolysis of C3 into C3H2O occurs in the alternative pathway. Cleavage of factor B (FB) by factor D yields Bb, which associates with C3H2O to form a C3 convertase. Cleavage of C3 and FB produces C3b and Bb, respectively. The binding of properdin to microbial surfaces recruits C3b, facilitating the assembly of the C3 convertase (C3bBb), and initiating pathway activation. Subsequent cleavage of C3 produces C3b, which combines with the C3 convertase to form a C5 convertase (C3bBbC3b). From this point, the terminal pathway is initiated to assemble the MAC, similarly to the classical and lectin pathways. Complement activation is regulated at various stages of the pathways by several membrane-bound complement regulatory proteins (Complement receptor 1 (CR1), CD46, CD55 and CD59) and circulating factors (C1-inhibitor (C1-INH), factor H (FH), factor I (FI), C4-binding protein (C4BP), clusterin and vitronectin), which are depicted in red, and properdin, which stabilises the alternative pathway C3 convertase.
2.2. Regulation of Complement Activation
Activation and amplification of the complement system induces a powerful inflammatory response, necessitating a regulatory system to avoid damage to host cells. This is achieved by a number of soluble and membrane-bound effector molecules which modulate various critical stages of the pathway, including the widely expressed membrane-bound complement regulatory proteins (mCRPs) (Table 1) and the fluid phase proteins C1 inhibitor, C4b-binding protein, factor H (FH) and factor I (FI) [35][48][49][35,48,49]. The alternative pathway C3 convertase is stabilised when bound by properdin [50].
Table 1.
Membrane-bound complement regulatory proteins.
| Regulator | Alternative Name (s) | Distribution | Function | Reference |
|---|---|---|---|---|
| CD35 | Complement receptor 1 (CR1) | Primarily lymphocytes, erythrocytes, phagocytes, dendritic cells | Cofactor for C3b and C4b degradation by Factor H Accelerates C3 and C5 convertases |
[51][52][53][54][55][51,52,53,54,55] |
| CD46 | Membrane cofactor protein (MCP) | All nucleated cells | Cofactor for C3b and C4b degradation by Factor H | [56][57][58][56,57,58] |
| CD55 | Decay accelerating factor (DAF) | Ubiquitously expressed | Accelerates decay of C3 and C5 convertases | [59][60][61][59,60,61] |
| CD59 | Membrane-inhibitor of reactive lysis (MIRL), MAC inhibitory protein (MAC-IP), Protectin | Ubiquitously expressed | Binds C5b-C9 to prevent polymerization of C9 | [62][63][62,63] |
2.3. Functions of Complement
MAC-induced lysis is the central cytotoxic event resulting from complement cascade activation, however, complement opsonins and anaphylatoxins also contribute to host defence. C3a and C5a exert their biological functions by binding to their respective receptors, the C3a receptor (C3aR) and the C5a receptor 1 (C5aR1/CD88), two G protein-coupled receptors [64][65][64,65]. A second, lesser understood C5aR, C5aR2 (previously C5L2) also exists [66]. In contrast, this 7-transmembrane receptor is uncoupled to G-proteins but is capable of recruiting β-arrestins [67][68][69][67,68,69]. C3a and C5a have been demonstrated to induce chemotaxis of mast cells [70][71][70,71] and eosinophils [72], with C5a also acting as a chemoattractant for macrophages [73], monocytes [74] neutrophils [74][75][74,75], basophils [76] and T and B lymphocytes [77][78][77,78]. Complement opsonins such as the C3 fragments C3b and iC3b aid phagocytosis by allowing recruited phagocytes to adhere to target cells via complement receptor 1 (CR1), complement receptor 3 (CR3), complement receptor 4 (CR4) and complement receptor Ig [4][79][4,79]. The phagocytic response to immune complexes may be enhanced by C5a-mediated upregulation of activating fragment crystallisation (Fc) γ receptors (FcγR) on the surface of phagocytes [24][80][24,80].
Complement cascade components also play key roles in orchestrating adaptive immunity. The complement receptors CR1 and CR2 are essential in the generation of B cell and follicular dendritic cell responses [81]. B cell responses are augmented by the binding of antigen opsonized by C3d to CR2, which leads to enhanced signalling through the B cell receptor and subsequent lowering of the threshold for activation [82][83][84][82,83,84]. In addition, complement components play roles in the priming and differentiation of T cells and provide survival signals to naïve T cells in an autocrine fashion [85][86][87][88][85,86,87,88]. Complement-mediated regulation of immune cells has been demonstrated within the TME [16][89][16,89]. The implication of these interactions on the efficacy of cancer treatment will be discussed later in this review.