β-cell mitochondria sense and shape calcium signals, linking the metabolism of glucose and other secretagogues to the generation of signals that promote insulin secretion during nutrient stimulation.
- mitochondria
- calcium
- insulin secretion
- β-cell
- diabetes
1. Introduction
Pancreatic β-cells constitute a small endocrine tissue organized, together with other endocrine cells, in the islets of Langerhans, scattered throughout the exocrine tissue of the pancreas [1]. β-cells sense glucose and secrete insulin in order to lower blood glucose levels after a meal. Defective insulin secretion underlies diabetes mellitus, which is a metabolic disorder characterized by elevated blood glucose levels [2][3][2,3]. The WHO’s first global report on diabetes indicates that the number of adults living with diabetes has almost quadrupled since 1980 to 422 million adults. This dramatic increase is largely due to the rise in Type 2 diabetes, whose driving factors include overweight and obesity. This disease develops when the β-cells of the endocrine pancreas fail to secrete sufficient hormones to compensate for the insulin resistance in the peripheral target tissues, liver, muscle and fat [4]. Diabetes is a non-communicable disease for which new approaches to prevention and treatment urgently need to be found. Targeting pancreatic β-cells is a promising strategy for the treatment of diabetes, due to the crucial role of the pancreatic β-cell in the pathogenesis of both Type 1 and Type 2 diabetes [5]. Therefore, preservation, expansion or improved function of β-cells are current approaches for targeting this cell type in the management of diabetes. Modulation of the biological pathways that regulate β-cell function represents the next stage of discovery in this field [5].
In this context, targeting mitochondrial Ca2+ represents an innovative approach to modulate β-cell function and to potentially promote beneficial effects for diabetic patients. Thus, dysregulation of Ca2+ signaling has been reported to have profound effects on β-cell performance and to increase the risk of developing diabetes [6][7][6,7]. Furthermore, modulation of dynamic cellular Ca2+ homeostasis has been proposed to prevent cytokine-mediated β-cell loss in diabetes [8]. In the pancreatic β-cell, Ca2+ homeostasis and β-cell function are substantially linked to mitochondrial function [9]. Therefore, mitochondria play a key role in β-cells during nutrient stimulation by linking the metabolism of glucose and other secretagogues to the generation of signals that promote insulin secretion [9]. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells [10]. Mitochondria are versatile intracellular organelles that are able to take up and release calcium [11][12][11,12]. Mitochondrial matrix Ca2+ is an activating signal for insulin secretion, and its requirements for signal-dependent hormone secretion have been highlighted [13]. Recently, the molecular identity of the mitochondrial Ca2+ uniporter (MCU), the transporter that mediates mitochondrial calcium uptake, has been revealed [14]. Genetic and pharmacological evidence has demonstrated the crucial role of mitochondrial Ca2+ in modulating pancreatic β-cell signal transduction, opening new perspectives for intervention [15][16][17][15,16,17].
2. Pancreatic β-Cell Signal Transduction and Ca2+ Homeostasis
In the pancreatic β-cell, metabolism–secretion coupling describes the molecular mechanism linking nutrient sensing and signaling to insulin secretion. This process relates to the consensus model and additional coupling factors (including both triggering and amplifying pathways) of glucose-stimulated insulin secretion [18][19][22,23]. Glucose-stimulated insulin secretion is relatively well characterized and requires the sequential activation of several biological processes (Figure 1).
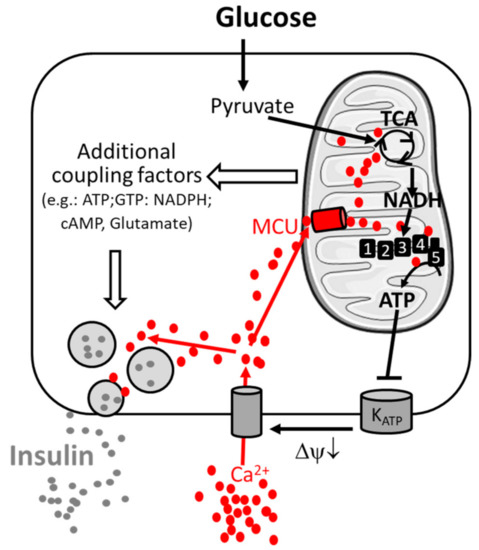
Figure 1. Consensus model of the signal transduction pathway of pancreatic β-cells. Metabolism–secretion coupling of β-cells requires the sequential activation of glycolysis, mitochondrial oxidative metabolism and Ca2+ entry through the plasma membrane. Glucose stimulates glycolysis and pyruvate production. Pyruvate triggers mitochondrial metabolism and the formation of the reduced form of nicotinamide adenine dinucleotide NADH (by the TCA cycle), which is the fuel for the respiratory complexes (1,2,3,4), enabling ATP production by ATP-synthase (5). ATP inhibits the KATP channel, inducing membrane depolarization (ΔΨ↓) and Ca2+ entry through voltage-gated Ca2+ channels, promoting insulin secretion. Ca2+ is taken up in parallel by mitochondria via the mitochondrial Ca2+ uniporter (MCU) and facilitates sustained insulin secretion. The amplifying pathway of metabolism–secretion coupling is co-generated by additive coupling factors. Additional players contributing to Ca2+ homeostasis are mentioned in the main text.
Glucose enters the β-cell by glucose-mediated transporters (GLUT). In the cytosol, it is metabolized by glycolysis to generate pyruvate, which is taken up by mitochondria. Mitochondrial pyruvate is metabolized by the tricarboxylic acid (TCA) cycle, which generates reducing equivalents NADH (reduced nicotinamide adenine dinucleotide) and FADH2 (reduced flavin adenine dinucleotide), which are substrates of the mitochondrial respiratory chain. Activation of mitochondrial respiration leads to mitochondrial ATP synthesis and thus to an increased cytosolic ATP-to-ADP ratio, which induces the closure of plasma membrane KATP channels and promotes plasma membrane depolarization. This opens voltage-gated plasma membrane Ca2+ channels, leading to an increase in cytosolic Ca2+ concentration, which finally triggers insulin exocytosis by activating Ca2+-sensitive granule-resident proteins (e.g., synaptotagmin-7 [20][24]). The amplifying pathways of metabolism–secretion coupling are contributed by additive coupling factors, and mitochondria have been characterized as a source of coupling factors [9][18][9,22].
The coupling between nutrient stimulation and hormone secretion is closely linked to Ca2+ homeostasis in the pancreatic β-cell [7][21][22][23][7,25,26,27]. Therefore, insulin secretion is driven by electrical activity and oscillations of intracellular Ca2+ concentrations. However, in addition to KATP channels and voltage-dependent Ca2+ channels [24][28], other plasma membrane channels and intracellular stores have been shown to be involved in insulin secretion in pancreatic β-cells [22][25][26,29]. In particular, store-operated Ca2+ channels, which are voltage-independent Ca2+ channels activated upon depletion of the endoplasmic reticulum Ca2+ stores, and transient receptor potential channel 1 (TRPC1), have been indicated to be involved in insulin secretion [26][27][30,31]. For store-operated Ca2+ channels, Orai1 has been identified as the main protein that conducts the previously described Ca2+ release-activated current (ICRAC). The activity of Orai1 channels is tightly controlled by the endoplasmic reticulum membrane protein stromal interacting molecule 1 (STIM1), which acts as an endoplasmic reticulum Ca2+ sensor and translocates, upon endoplasmic reticulum depletion, to endoplasmic reticulum/plasma membrane regions, where Orai1 is clustered [27][31]. In addition, mobilization of intracellular Ca2+ from the endoplasmic reticulum has been suggested to potentiate glucose-stimulated hormone secretion [28][32], and the Type 2 ryanodine receptor (RyR2) has been proposed to play a crucial role in regulating insulin secretion and glucose homeostasis [29][33]. Moreover, an atypical Ca2+ leak has been observed in the endoplasmic reticulum, specifically in pancreatic islets and β-cells. This continuous Ca2+ efflux from the endoplasmic reticulum was modulated by GSK3β-dependent phosphorylation of presenilin-1 and promoted mitochondrial activation [30][31][34,35]. Additional intracellular acidic compartments may contribute to the local modulation of β-cell Ca2+ homeostasis and thus β-cell function, including insulin granules and other acidic stores (e.g., lysosomes) [22][26].
The last player in pancreatic β-cell Ca2+ homeostasis and signal transduction is represented by the mitochondrial network. Therefore, mitochondria are intracellular organelles that take up and release Ca2+, promoting the sensing and shaping of cytosolic Ca2+ signals [32][36]. The role of mitochondrial Ca2+ signaling in energized mitochondria of the pancreatic β-cell is emerging as a biological process of critical importance to pancreatic β-cell function and is highlighted in the next section.
3. The Mitochondrial Calcium Uniporter and Its Existence in Pancreatic β-Cells
Despite the undisputed role of cytosolic Ca2+ elevation in triggering insulin secretion in the pancreatic β-cell, it is also accepted that such a rise in itself does not sustain insulin secretion [33][34][35][18,37,38]. Therefore, mitochondria have been demonstrated to contribute to robust insulin secretion by triggering additional regulatory factors, and it has been proposed that mitochondrial Ca2+ plays a crucial role as a receiver and generator of the signals essential for metabolism–secretion coupling [9][36][9,39]. The discovery and molecular definition of the MCU [14][37][14,40], the transporter which mediates the transport of Ca2+ in the mitochondrial matrix under physiological conditions, is shedding light on the role of mitochondrial Ca2+ elevation in different tissues, including the pancreatic β-cell (see Section 4).
The MCU complex is a low-affinity, high-capacity Ca2+ uniporter embedded in the inner mitochondrial membrane with an approximate in vitro Ca2+ binding affinity estimated to be between 10 and 70 µM [38][39][40][41][41,42,43,44] in different tissues. The transport of Ca2+ ions into the mitochondrial matrix is electrochemically driven by a strong electrical gradient (~180 mV) through the ion-impermeable inner mitochondrial membrane [42][45]. As reported for many other tissues, in the pancreatic β-cell, resting-state mitochondrial Ca2+ levels are also very close to cytosolic Ca2+ levels, indicating tight regulation of the MCU [33][18]. Permeabilized INS-1 cells (a cell-line model of pancreatic insulin-secreting cells) display significant mitochondrial Ca2+ uptake when perfused with buffers containing a Ca2+ concentration of only 150 nM [43][46], which is below the general threshold of MCU activation [44][47]. This study suggests a slightly higher affinity configuration of the β-cell MCU, compared with the MCU of other tissues. However, in permeabilized β-cells perfused with a range of different Ca2+ buffers, mitochondrial Ca2+ uptake was more efficient above 2 µM [45][48]. These data indicate that although some studies demonstrated a lower Ca2+ activation threshold, the MCU of β-cells also behaves as a low affinity, high capacity Ca2+ transport system, with similar properties to those reported in other tissues. In any case, the mitochondrial Ca2+ concentration in glucose-stimulated β-cells was reported to reach only 600–800 nM [13][33][46][13,18,49], mirroring cytosolic Ca2+ events. Purinergic activation and potassium-induced depolarization generate cytosolic Ca2+ transients around one micromolar, with the corresponding mitochondrial Ca2+ rises reaching nearly 5 µM [36][39]. These results indicate that, on average, β-cell mitochondria barely reach micromolar Ca2+ concentrations in the matrix during physiological activation.
The MCU complex consists of at least six subunits, each of which plays an individual role in orchestrating mitochondrial Ca2+ uptake [38][41]. The MCU subunit is a 40 kDa protein that, together with its paralog, MCUb [47][50], has been proposed to form a tetramerizing pore which penetrates the inner mitochondrial membrane [48][51]. In order to channel Ca2+ ions into the mitochondrial matrix, it requires a third subunit known as the essential MCU regulator (EMRE), a 10 kDa protein [47][49][50,52]. EMRE is a single-transmembrane protein whose transmembrane helix connects it to the MCU subunit [50][53]. The MCU and MCUb share 50% sequence similarity and have opposite effects on mitochondrial Ca2+ uptake [47][50]. While the MCU has a promoting effect on mitochondrial Ca2+ uptake, MCUb exerts a negative effect on mitochondrial Ca2+ uptake [51][54]. The ratio of MCU and MCUb varies among tissues and is potentially influenced by metabolic impairments, leading to the hypothesis that an altered MCU/MCUb ratio may affect β-cell function [52][53][54][55,56,57]. When Ca2+ in the intermembrane space exceeds ~0.6 µM, it initiates activation of the MCU complex via the MCU gatekeeper protein paralogs mitochondrial calcium uptake 1 and 2 (MICU1 and MICU2) [41][55][44,58]. MICU2 is a 50 kDa protein and shares approximately 25% sequence identity with the 54 kDa protein MICU1 [39][42]. Neither MICU1 nor MICU2 contain transmembrane domains that would link them to the MCU subunit, so it has been proposed that MICU1 is linked to the MCU via EMRE through electrostatic interaction [55][58]. However, a more recent study revealed an additional direct binding of MICU1 to the highly conserved DIME motif of the MCU [56][57][59,60]. In addition, another study showed an interaction between MICU1/MICU2 and cardiolipin [55][58]. Through cysteine residues, MICU2 forms an intermembrane space-facing heterodimer with MICU1 via disulfide bonds [48][51]. Both gatekeeper proteins contain EF-hand motifs (consisting of a helix E, a loop and another helix F), with a Ca2+ binding affinity of ~0.3 µM for MICU1 and a Ca2+ binding affinity of ~0.6 µM for the MICU1–MICU2 dimer [55][58].
Genetic ablation of MICU2 lowers the required Ca2+ concentration in the intermembrane space for MICU1-regulated MCU activation, resulting in increased mitochondrial Ca2+ uptake [49][52]. On the other hand, ablation of MICU1 prevents indirect binding of MICU2 to the MCU, resulting in unregulated Ca2+ entry into the mitochondria [58][59][61,62]. Studies with MICU1 knockout as well as with MICU1 knockdown have described an inverse relationship between extra-mitochondrial Ca2+ concentrations and mitochondrial Ca2+ uptake [59][60][62,63]. This suggests that the absence of MICU1 at low/resting extra-mitochondrial Ca2+ concentrations leads to increased mitochondrial Ca2+ uptake, while high extra-mitochondrial Ca2+ concentrations are associated with decreased mitochondrial Ca2+ uptake. The final subunit, which has been proposed to contribute to the structure of the MCU complex is a scaffolding factor called mitochondrial calcium uniporter regulator 1 (MCUR1), which can interact with the MCU but not with MICU1 [61][64]. However, there are two possible interpretations of the direct or indirect role of MCUR1 in modulating mitochondrial Ca2+ uptake. Several studies have demonstrated a regulatory effect of MCUR1 on MCU complex activity, noting reduced mitochondrial Ca2+ uptake in the absence of MCUR1 and a decrease in reducing equivalents as well as ATP generation [62][63][64][65,66,67]. Alternatively, MCUR1 has been suggested to have a potential role as a Complex IV assembly factor in the mitochondrial respiratory chain [65][68]. Interestingly, the relative abundance of distinct MCU components has been reported to be different in distinct cell types or tissues [31][35], and it has been proposed that the specific stoichiometry of the MCU subunits may define the functional characteristics of the channel, including Ca2+ permeation across the pore, the activation threshold and the cooperativity. Consistent with this hypothesis, it has been reported that in parallel with the distinct relative abundance of MCU components [31][35], MCU activity also varies greatly among cell types [66][69]. Although the stoichiometry of the different MCU subunits and their relative abundance in different tissues, including in the pancreatic β-cell, are largely unknown, we tentatively speculate that these cells may have a specific configuration of the MCU that reduces the Ca2+ activation threshold.
Pancreatic β-cells have been demonstrated to express a functional MCU, and some studies have investigated the role of MCU subunits on insulin secretion [15][16][17][15,16,17] (Figure 2).

Figure 2. The mitochondrial calcium uniporter (MCU) comprises distinct pore-forming (MCU/MCUb) and associated proteins (essential MCU regulator (EMRE), mitochondrial calcium uptake (MICU) 1, MICU2, MICU3 and mitochondrial calcium uniporter regulator 1 (MCUR1)). The subunits that have already been studied in models of the pancreatic β-cell are rendered in green. (A) Model of the closed conformation of MCU. (B) Following glucose stimulation, the entry of Ca2+ into the matrix space is mediated by MCU. IMM, inner mitochondrial membrane; IMS, intermembrane space.
Not surprisingly, the existence of MCU and MICU1 subunits in pancreatic β-cell models has been reported and functionally validated (see [15][16][17][52]Section 4) [15,16,17,55]. In addition, a study recently published as a preprint in bioRxiv but not yet peer-reviewed proposes the existence of MICU2 subunits in rat and human insulin-secreting cell lines as well as in mouse islets (Mitochondrial Clearance of Ca2+ Controls Insulin Secretion; https://doi.org/10.1101/830323 (accessed on 1 February 2021); Vishnu, Hamilton, Bagge, Wernersson, Cowan, Barnard, Sancak, Kamer, Spégel, Fex, Tengholm, Mootha, Nicholls and Mulder). Further studies are expected to consolidate the specific role of the other subunits of the MCU in the pancreatic β-cell (Figure 2) and fully define the structure of this transporter. However, the crucial role of the MCU complex in the mitochondrial Ca2+ transport of the pancreatic β-cell and in the pancreatic β-cell metabolism–secretion coupling is already well established and will be discussed in the next section.
(References would be added automatically after the entry is online)