The tripartite motif (TRIM) family comprises at least 80 members in humans, with most having ubiquitin or SUMO E3 ligase activity conferred by their N-terminal RING domain. TRIMs regulate a wide range of processes in ubiquitination- or sumoylation-dependent manners in most cases, and fewer as adaptors. Their roles in the regulation of viral infections, autophagy, cell cycle progression, DNA damage and other stress responses, and carcinogenesis are being increasingly appreciated, and their E3 ligase activities are attractive targets for developing specific immunotherapeutic strategies for immune diseases and cancers.
- TRIMs
- ubiquitination
- PRR
- IFN-I
- IRFs
1. Introduction
In mammalians, interferons (IFNs) include three types, type I, II, and III. Type I IFNs (IFN-Is) include the majority of 26 isoforms of IFNα that are encoded by 13 genes, and one IFNβ that is encoded by the single gene IFNB, as well as other minor subtypes, including IFNε, IFNκ, IFNω, IFNδ, IFNτ, and IFNζ. IFNαs are mainly secreted by plasmacytoid dendritic cells (pDCs) and IFNβ is mainly secreted by fibroblasts. All IFN-Is signal through the integral membrane IFNAR1 and -2 heterodimer, and play crucial roles in the first line of innate immune response and subsequent adaptive immune response in response to viral or bacterial infections [1].
Importantly, recent studies have shown that IFN-Is play a dual role in chronic viral infections. At the early stage of infection, they have potent antiviral activity. However, at late stages, a low level of prolonged IFN-I signaling, exemplified by chronic infection of viruses such as HIV and HCV [2][3][4][2,3,4], triggers long-term chronic immune activation that proceeds to T cell exhaustion and inflammaging/immunosenescence in both direct and indirect manners [5][6][5,6] and therefore serves as a bridge that links innate and adaptive immune responses [4][5][7][8][9][10][4,5,7,8,9,10]. For example, the engagement of TLR7 in HIV-infected CD4+ T cells induces anergy/unresponsiveness, accounting for the impaired T cell function by chronic HIV infection [11]. A prolonged IFN-I response also facilitates the establishment of TME (tumor microenvironment) [4][5][7][12][13][14][4,5,7,12,13,14]. IFN-Is also play crucial roles in cellular development and homeostasis [5][6][15][16][17][5,6,15,16,17]. Aberrant production of IFN-Is is associated with many types of diseases, including autoimmune disorders and cancers [6][18][19][20][6,18,19,20]. Therefore, it is of fundamental importance to understand the precise mechanisms of how IFN-Is are regulated in different biological contexts [21][22][21,22].
Ubiquitination is a pervasive theme equally important to phosphorylation of proteins in myriad processes. Ubiquitin (Ub) is a 76-amino acid protein that is ubiquitously distributed and highly conserved throughout eukaryotic organisms. The Ub protein can be free or conjugated to a lysine site of a protein substrate through its 3’-end. This conjugation process involves E1 activating enzyme, E2 conjugating enzyme, and E3 ligase, with the E3 ligase determining the specificity of the substrate. Ub itself has seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63), and each can serve as the Ub target to link another Ub. If only a single Ub is conjugated to each lysine site of the substrates, it is called mono (also only one lysine site on the substrate) or multi (more than one lysine site on the substrate) ubiquitination. If the substrate is Ub itself, polyubiquitin chains will be formed on the substrate. Usually, a polyubiquitin chain contains more than 4 Ub molecules. In the last decade, non-canonical ubiquitination types on serine, threonine, and cysteine sites other than lysine site have been identified, and their importance in specific cellular functions has been recognized [23][24][23,24].
The most well-understood type of ubiquitination is K48-linked polyubiquitination, which is principally known as the major process whereby proteins are targeted for proteasomal degradation through the 26S proteasome. Later, nonproteolytic types of polyubiquitination (represented by K63-linked polyubiquitination), monoubiquitination, and linear ubiquitination have been gradually identified [25][26][27][25,26,27]. More recently, other ubiquitination-like modifications (e.g., sumoylation, acetylation, ISGylation, neddylation, palmitoylation, and UFMylation) have also been discovered. The roles of these posttranslational modifications (PTMs) in a myriad of cellular processes, such as receptor internalization (endocytosis), vesicle trafficking, immune response and inflammation, DNA damage response, autophagy, and cell death, have been greatly appreciated [27][28][29][30][31][32][33][34][35][27,28,29,30,31,32,33,34,35].
IFN-I production is controlled at multiple layers to ensure appropriate mounting of antiviral and antitumor immune responses. It is clear that both host and viral ubiquitin systems play pivotal roles in IFN-I-mediated innate immunity and in cellular transformation mediated by oncogenic viruses represented by EBV (Epstein-Barr Virus), KSHV (Kaposi’s sarcoma-associated herpesvirus), and HPV (human papillomavirus) [36][37][38][39][40][41][36,37,38,39,40,41].
2. PRR Signaling Pathways to IFN-I Production
IFN-Is are produced downstream of the signaling pathways of host germline-encoded pathogen recognition receptors (PRRs), which are expressed on the cell membrane or in the cytoplasm of the cells of the innate immune system, in response to pathogen-associated molecular patterns (PAMPs) that include pathogenic nucleic acids, LPS, and proteins, or in response to host damage-associated molecular patterns (DAMPs), such as self-nuclei acids, heat-shock proteins, and HMGB1. Recognition of PAMPs or DAMPs by PRRs triggers signal cascades that activate the transcription factors, including NFκB, Interferon regulatory factors (IRFs), and AP1, or activate caspase-mediated cell death and inflammation.
PRRs include the well-known transmembrane Toll-like receptors (TLRs) (Figure 1) and an increasing pool of “Toll-free” receptors [22]. Endosomal TLRs (TLR3, -7, -9, and murine TLR8) and endocytic TLR4, as well as cytoplasmic RIG-I and cGAS, amongst others, are able to recognize pathogenic or host cell nucleic acids (LPS for TLR4) to activate IRFs in addition to NFκB and AP1, which induce IFN-Is and also pro-inflammatory cytokines [42]. Self-nucleic acids are derived from the nucleus or mitochondria of the cells suffering from endogenous or exogenous stresses, such as DNA replication, oxidative stress, DNA damage, and cell death [43][44][45][46][47][43,44,45,46,47]. While the transcription of IFNαs is solely dependent on IRFs, full transactivation of the IFNβ promoter requires the cooperation of IRFs, NFκB, and other co-factors in the transcriptional complex named enhanceosome [48].
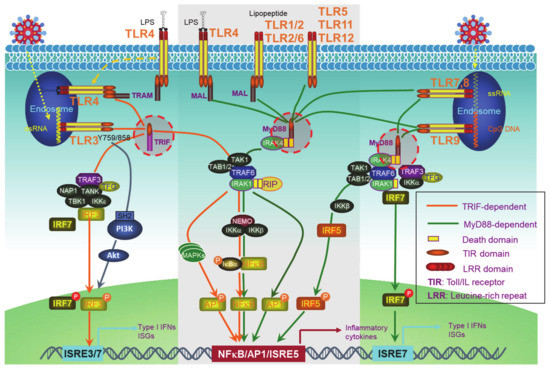
Figure 1. Toll-like receptor (TLR)signaling pathways. The TLR family has 13 members, among which, endosomal TLR3, -7, -9, and murine TLR8, and endocytic TLR4 are able to trigger signaling for IFN-I production. TLRs that cannot trigger the activation of Interferon regulatory factors (IRFs) (The middle part in the gray frame) do not contribute to IFN-I production. All the TLRs have a TIR domain in the cytoplasm, which recruits adaptor proteins also with a TIR domain at their C-terminus. TRIF and MyD88, two adaptor proteins, bridge all TLRs to downstream signaling molecules, leading to the activation of NFκB, IRFs, and AP1. IRF1, -3, -5, -7, and -8 are the transcription factors for both IFNα and IFNβ transcription in different cell contexts, but a full IFNβ transcription requires the enhanceosome complex that contains NFκB, IRF3, -7, ATF-2/c-Jun, and HMGIY (high mobility group I(Y)). ISRE: Interferon-stimulated response element. ISRE3/7: ISRE that binds to IRF3//7. ISRE7: ISRE that binds to IRF7.
3. The TRIM Family
The tripartite motif (TRIM) family of proteins is large and includes at least 80 members in humans, with most having E3 ligase activity for target-specific ubiquitination, and plays crucial roles in innate immunity, transcription, autophagy, and carcinogenesis [49][50][49,50]. The N-terminal TRIM motif includes the conserved RBCC domain that comprises of three subdomains: 1 RING domain that confers with E3 ligase activity (8 human TRIMs do not have the RING domain), 0~2 B-box ZNF domains (B1+B2 or B2 alone), and 0~1 coil–coil region that is associated with B-boxes. According to the diversity of the C-terminuses and genomic organization, TRIM proteins are grouped into Group1 and Group 2. Members in Group 1 possess a variety of C-terminal domains (COS, FN3, ACID, PRY, PHD-BROMO, FIL, NHL, MATH, ARF, and TM) and exist in both vertebrate and invertebrates, and those in Group 2 possess a C-terminal SPRY domain, and they are absent in invertebrates (Figure 2) [49][50][51][49,50,51]. The SPY-SPRY domain is critical for TRIM proteins’ interaction with their substrates.
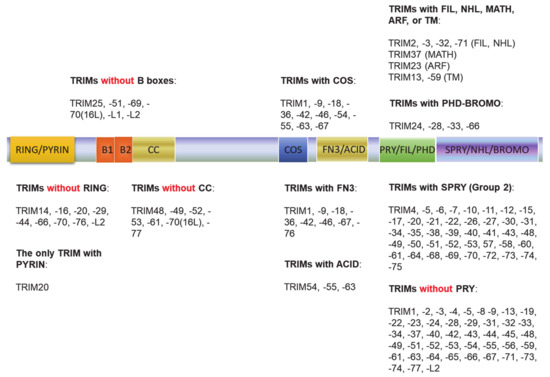
Figure 2. The tripartite motif (TRIM) family protein domain alignment. The TRIM family includes at least 80 members in humans. The N-terminal TRIM motif includes the conserved RBCC domain that comprises of three subdomains: 1 RING domain that confers with E3 ligase activity (8 TRIMs in humans do not have the RING domain), 0~2 B-box ZNF domains (B1+B2 or B2 alone), and 0~1 coil–coil region that is associated with B-boxes. According to the diversity of the C-terminuses and genomic organization, TRIM proteins are grouped into Group1 and Group 2. Members in Group 1 possess a variety of C-terminal domains (COS, FN3, ACID, PRY, PHD-BROMO, FIL, NHL, MATH, ARF, and TM) and exist in both vertebrate and invertebrates, and those in Group 2 possess a C-terminal SPRY domain, and they are absent in invertebrates.
Many TRIMs are inducible by IFN-Is, and play crucial roles in IFN-I-mediated innate immune regulation, with the involvement of ubiquitination and sumoylation in most cases [50][52][53][54][55][56][57][58][50,52,53,54,55,56,57,58]. These TRIMs can target most if not all components of the PRR and Jak-STAT1 IFN-I pathways, including different ligands (PAMPs and DAMPs); the receptors such as TLRs, cGAS, and DDX41; the adaptors MyD88, TRIF, STING, and TRAF6 and -3; the kinases IKKs and TAK1; and the transcription factors IRF3 and -7 and NFκB (Table 1). TRIM genes evolve parallelly with the immune system, further supporting their roles as regulators of immune responses [51][53][51,53].
Table 1. TRIMs in the regulation of Interferon (IFN)-I-mediated innate immune network.
TRIM | Synonym | Targets in the IFN-I Network | Ub Conjugation Type | Outcome of Conjugation | Selected References | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
TRIM3 | RNF97 | TLR3 | K63 | Promotes ESCRT-mediated TLR3 sorting to endosomes |
[59] |
||||||||||||||||||
TRIM4 | RNF87 | RIG-I | K63 | Activation |
[60] |
||||||||||||||||||
TRIM5α | RNF88 | HIV Gag | K48 | Degradation | |||||||||||||||||||
TAK1 | K63 | Activation |
[63] |
||||||||||||||||||||
TRIM12c in mice | TRAF6 | K63 (?) | Activation |
[62] |
|||||||||||||||||||
TRIM6 | RNF89 | Ebola VP35 | Poly | Promotes VP35 IFN-I inhibitory activity |
[64] |
||||||||||||||||||
IKKε | Free K48 | Activation of IKKε, leading to STAT1 activation |
[65] |
||||||||||||||||||||
TRIM7 | RNF90 | GNIP | Zika virus envelope (E) | K63 | Enhances virus attachment and entry into the cell |
[66] |
|||||||||||||||||
TLR4 | NA* | Promotes TLR4 activation |
[67] |
||||||||||||||||||||
TRIM8 | RNF27 | GERP | TRIF | K6, K33 | Disrupts the TRIF-TBK1 complex |
[68] |
|||||||||||||||||
TAK1 | K63 | Activation | |||||||||||||||||||||
IRF7 | Protects p-IRF7 from Pin1-mediated proteasomal degradation in the nucleus |
[71] |
|||||||||||||||||||||
SOCS1 | K48 (?) | Degradation |
[72] |
||||||||||||||||||||
PIAS3 | K48 | Degradation |
[73] |
||||||||||||||||||||
Interaction (?) | Promotes PIAS3 nucleus-to-cytoplasm translocation |
[74] |
|||||||||||||||||||||
TRIM9s | RNF91 | SPRING | TBK1 | Interaction | Recruits GSK3β and TBK1, leading to TBK1 activation |
[75] |
|||||||||||||||||
TRIM9 | β-TrCP | Interaction | Stabilizes IκBα | ||||||||||||||||||||
TRIM11 | RNF92 | BIA1 | TBK1 | Interaction | Inhibits TBK1 activation |
[77] |
|||||||||||||||||
TRIM5 | NA* | Degradation |
[78] |
||||||||||||||||||||
TRIM13 | RNF77 | RFP2 | CAR | LEU5 | DLEU5 | RIG-I | Interaction | Potentiates RIG-I activity |
[79] |
||||||||||||||
MDA5 | Interaction | Inhibition |
[79] |
||||||||||||||||||||
TRAF6 | K29 | Activation |
[80] |
||||||||||||||||||||
NEMO | K48 | Degradation |
[81] |
||||||||||||||||||||
TRIM14 | KIAA0129 | HCV NS5A | K48 (?) | Degradation |
[82] |
||||||||||||||||||
cGAS, TBK1 | Interaction | Inhibition of autophagic degradation of cGAS | |||||||||||||||||||||
MAVS | Interaction | Recruitment of NEMO to MAVS signalosome |
[84] |
||||||||||||||||||||
TRIM15 | RNF93 | ZNF178 | ZNFB7 | MAVS | NA* | Promotes RIG-I-mediated IFN production |
[86] |
||||||||||||||||
TRIM19 | RNF71 | PML | MYL | HIV genome | Sequestrates HIV genome in the cytoplasm, blocking HIV transduction |
[87] |
|||||||||||||||||
HFV Tas | Represses HFV transcription by preventing Tas binding to viral DNA |
[88] |
|||||||||||||||||||||
LCMV Z | Inhibits LCMV replication |
[89] |
|||||||||||||||||||||
hCMV IE1 | Interaction | IE1 forms a complex with TRIM19-STAT1/2 to impede IFN-I signaling |
[90] |
||||||||||||||||||||
STAT1/2 | Induction and stabilization, promoting IFN-I signaling |
[90] |
|||||||||||||||||||||
Pin1 (by TRIM19IV) | Regulates the cellular distribution of Pin1 |
[91] |
|||||||||||||||||||||
Ubc9 (The only SUMO E2) | Required for IFN-induced global sumoylation |
[92] |
|||||||||||||||||||||
NFκB | Inhibits NFκB-mediated transcription and survival |
[93] |
|||||||||||||||||||||
Promotes IKKε-mediated p65 phosphorylation and NFκB activity |
[94] |
||||||||||||||||||||||
ROS | Functions as an ROS sensor promoting p53 activation |
[95] |
|||||||||||||||||||||
TRIM20 | Pyrin | MEFV | p65 | Interaction | Promotes p65 nuclear translocation |
[96] |
|||||||||||||||||
IκBα | Promotes IκBα degradation |
[96] |
|||||||||||||||||||||
TRIM21 | RNF81 | Ro52 | SSA1 | DDX41 | K48 | Degradation |
[97] |
||||||||||||||||
MAVS | K27 | Activation |
[98] |
||||||||||||||||||||
FADD | Interaction | Promotes IRF7 ubiquitination-mediated degradation |
[99] |
||||||||||||||||||||
TAK1 | Free K63 | Activates TAK1, leading to the activation of NFκB, AP1, and IRFs | |||||||||||||||||||||
IKKβ | Mono-Ub | Autophagic degradation |
[102] |
||||||||||||||||||||
IRF3 | Interaction | Protects p-IRF3 from Pin1-mediated proteasomal degradation |
[103] |
||||||||||||||||||||
K48 | Targets IRF3 for proteosomal degradation | ||||||||||||||||||||||
Interacts with ULK1, Beclin1, and p62 | Targets IRF3 for autophagic degradation |
[106] |
|||||||||||||||||||||
IRF5 | Various | Degradation of isoforms V1 and V5, but not V2 or V3 |
[107] |
||||||||||||||||||||
IRF7 | K48 | Degradation |
[108] |
||||||||||||||||||||
IRF8 | NA* | Activation |
[109] |
||||||||||||||||||||
TRIM22 | RNF94 | STAF50 | HIV Gag, LTR | Degradation |
[110] |
||||||||||||||||||
Influenza A Virus NP | Degradation |
[111] |
|||||||||||||||||||||
HCV NS5A | K48 (?) | Degradation |
[112] |
||||||||||||||||||||
TAB2 | K48 (?) | Degradation |
[113] |
||||||||||||||||||||
TRIM23 | RNF46 | ARD1 | ARFD1 | TRAF3 | Interaction | Function not clear, likely promoting TRAF3-mediated antiviral activity |
[114] |
||||||||||||||||
TRAF6 | Interaction | Activation of NFκB mediated by HCMV UL144 |
[115] |
||||||||||||||||||||
NEMO | K27 | Activation |
[114] |
||||||||||||||||||||
TBK1 | K27 of TRIM23 (self) | Recruits and activates TBK1, inducing TBK1-mediated autophagy |
[116] |
||||||||||||||||||||
TRIM24 | RNF82 | TIF1A | TRAF3 | K63 | Activation |
[117] |
|||||||||||||||||
RARα | Interaction | Inhibits RARα activity and retinoic acid-induced STAT1 expression |
[118] |
||||||||||||||||||||
p53 | K48 (?) | Promotes p53 ubiquitination and degradation |
[119] |
||||||||||||||||||||
TRIM25 | RNF147 | ZNF147 | Influenza virus vRNP | Blocks vRNA chain elongation |
[120] |
||||||||||||||||||
RIG-I | K63 | Activation | |||||||||||||||||||||
MAVS | K48 | Degradation |
[123] |
||||||||||||||||||||
ISG15 | Functions as an ISG15 E3 ligase |
[124] |
|||||||||||||||||||||
ZAP | K48, K63 | Critical for ZAP inhibition of viral genome translation |
[125] |
||||||||||||||||||||
TRIM26 | RNF95 | ZNF173 | AFP | TBK1 | K27 of TRIM26 (self) | Bridges TBK1-NEMO interaction, leading to TBK1 activation |
[126] |
||||||||||||||||
IRF3 | K48 | Degradation |
[127] |
||||||||||||||||||||
TRIM27 | RNF76 | RFP | TBK1 | K48 | Degradation | ||||||||||||||||||
IKKα, IKKβ, IKKε | Interaction | Inhibition |
[131] |
||||||||||||||||||||
TRIM28 | RNF96 | KAP1 | IRF7 | Sumoylation | Inhibition |
[132] |
|||||||||||||||||
TRIM29 | ATDC | STING | K48 | Degradation | |||||||||||||||||||
MAVS | K11 | Degradation |
[135] |
||||||||||||||||||||
NEMO | K48 | Degradation |
[136] |
||||||||||||||||||||
TRIM30α | RPT1 | STING | K48 | Degradation |
[137] |
||||||||||||||||||
TAB2/3 | Lysosomal degradation |
[138] |
|||||||||||||||||||||
TRIM31 | RNF | HCG1 | MAVS | K63 | Promotes MAVS signalosome assembly |
[139] |
|||||||||||||||||
TRIM32 | TATIP | BBS11 | HT2A | Influenza PB1 | K48 | Degradation |
[140] |
||||||||||||||||
STING | K63 | Activation |
[141] |
||||||||||||||||||||
TRIF | NA* | Targets TRIF for TAX1BP1-mediated autophagic degradation |
[142] |
||||||||||||||||||||
TRIM33 | TIF1γ | HIV integrase | K48 | Degradation |
[143] |
||||||||||||||||||
TRIM35 | HLS5 | MAIR | TRAF3 | K63 | Activation |
[144] |
|||||||||||||||||
IRF7 | K48 | Degradation |
[145] |
||||||||||||||||||||
TRIM38 | RNF15 | RORET | RIG-I, MDA5 | Sumoylation | Stabilization |
[146] |
|||||||||||||||||
cGAS, STING | Sumoylation | Stabilization |
[147] |
||||||||||||||||||||
TRAF6 | K48 | Degradation |
[148] |
||||||||||||||||||||
NAP1 | K48 | Degradation |
[149] |
||||||||||||||||||||
TAB2 | K48? | Degradation |
[150] |
||||||||||||||||||||
TRIF | K48 | Degradation | |||||||||||||||||||||
TRIM39 | RNF23 | TFP | Cactin | NA* | Stabilizes Cactin, inhibiting NFκB and IRFs |
[152] |
|||||||||||||||||
TRIM40 | RNF35 | RIG-I, MDA5 | K27, K48 | Degradation |
[153] |
||||||||||||||||||
NEMO | Neddylation | Inhibition |
[154] |
||||||||||||||||||||
TRIM41 | RINCK | MGC1127 | cGAS | Mono-Ub | Activation |
[155] |
|||||||||||||||||
TRIM44 | DIPB | AN3 | MAVS | Interaction | Stabilization of MAVS by preventing its ubiquitination |
[156] |
|||||||||||||||||
TRIM45 | RNF99 | NFκB | E3 ligase activity not required | Inhibition of TNFα-mediated NFκB activation |
[157] |
||||||||||||||||||
TRIM56 | RNF109 | Influenza virus RNA | Inhibits vRNA synthesis |
[158] |
|||||||||||||||||||
cGAS | Mono-Ub | Activation |
[159] |
||||||||||||||||||||
STING | K63 | Activation |
[160] |
||||||||||||||||||||
TRIF | Interaction | Activation |
[161] |
||||||||||||||||||||
TRIM59 | RNF104 | TSBF1 | MRF1 | IFT80L | ECSIT | Interaction | Inhibition of TLR singling pathways to activate NFκB and IRFs |
[162] |
|||||||||||||||
TRIM62 | DEAR1 | TRIF | NA* | Activation |
[86] |
||||||||||||||||||
TRIM65 | MDA5 | K63 | Activation | ||||||||||||||||||||
TRIM68 | RNF137 | SS56 | TFG | various | Induces TFG lysosomal degradation |
[163] |
|||||||||||||||||
* NA: not assayed. Question marks (?) refer to “very likely but not experimentally revealed”.
4. TRIMs in Regulating PRR Signaling Pathways to IFN-I Production
Upon binding to PAMPs or DAMPs, PRRs trigger signals that transmit via unique adaptors to the Ub E3 ligase TRAF6 or -3 and then orchestrate to activate the kinase cascades IKKs, IRAKs, and MAPKs for the activation of the transcription factors NFκB, IRFs, and AP1. Ubiquitination regulates the cellular trafficking, stability, complex assembly, and activity of different components in PRR signaling cascades (Figure 3).
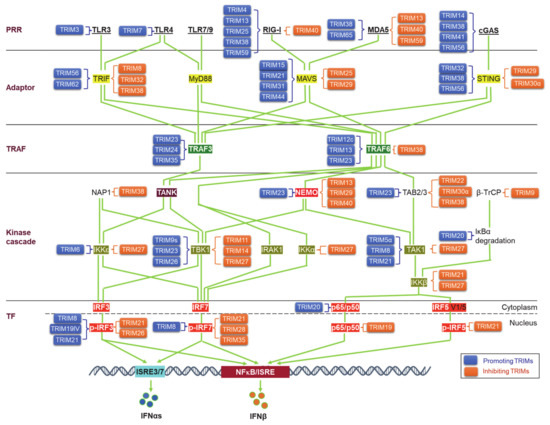
Figure 3. TRIM regulation of pathogen recognition receptor (PRR) pathways. TRIMs are involved in the regulation of the stability and activity of PRR components in all cascades of the signaling pathways, including ligands, receptors, adaptors, TRAFs, kinases and associated regulators, and the final transcription factors (TFs). An increasing pool of regulatory factors of the PRR pathways is also regulated by TRIMs (not shown). TRIMs promoting the stability or activity of the targets are shown on the left of the targets (blue), and those inhibiting the targets are shown on the right of the targets (brown). TRIM19IV and TRIM21 positively regulate phosphorylated IRF3 in indirect manners via Pin1. As such, TRIM8 positively regulates phosphorylated IRF7 in an indirect manner via Pin1. Other indirect regulations of these PRR pathways by TRIMs are not shown. TF: Transcription factor.
Viral PAMPs and other viral components can be targeted by the host Ub system, including a subset of TRIMs, for ubiquitination-mediated degradation in most cases, and thus the IFN-I response is blocked at the very beginning to suppress viral replication, with some examples listed in Table 1 [54][58][54,58]. Of note, TRIM5α (also TRIM22) targets HIV1 Gag and plays a unique role in restricting HIV1 infection (and other retroviruses), implicating a potential clinical application [61][62][61,62]. In fewer cases, TRIMs can promote viral entry and replication by targeting viral proteins. For example, TRIM7 targets Zika virus envelope protein E for K63-linked ubiquitination that enhances viral attachment to the cell surface and promotes viral entry [66]. VP35, the Ebola virus polymerase co-factor, has IFN-I inhibitory activity. TRIM6 promotes VP35 polyubiquitination to enhance viral infection [64].
5. TRIMs in Regulating the Jak-STAT IFN-I Signaling
The level of initial IFN-Is produced downstream of PRR pathways upon viral infection is relatively low due to the low level of endogenous IRF7 protein; these priming IFN-Is then secret to outside of the cell in autocrine and paracrine manners, and bind to IFN-I receptor (IFNAR) on other cells, consequently triggering the Jak-STAT IFN-I pathway, which serves as the second phase of antiviral response by inducing the expression of more IRF7, which in turn participates in IFN-I production downstream of PRR signaling, therefore amplifying the IFN-I production in a positive regulatory circuit (Figure 4) [42].
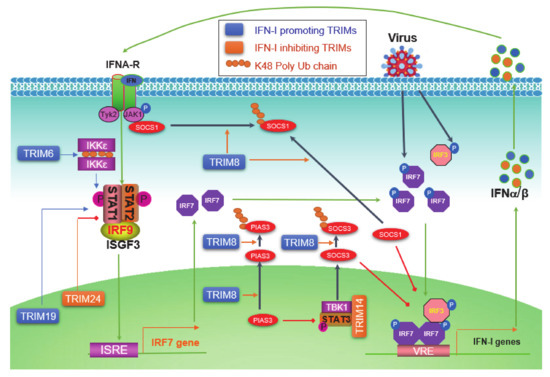
Figure 4.
Jak-STAT pathways are well known to be negatively regulated by two families: SOCS (Suppressor of cytokine signaling) and PIAS (Protein inhibitor of activated STAT). TRIM8 can shuttle between the cytoplasm and the nucleus [74], and has multiple functions to promote IFN-I signaling. TRIM8 promotes proteasomal degradation of SOCS1 and PIAS3 presumedly in the cytoplasm, and also nuclear TRIM8 promotes PIAS3 nucleus-cytoplasm translocation to inhibit PIAS3 activity [72][73][74][72,73,74]. SOCS1 not only inhibits Jak1 activity by directly binding to phosphorylated Jak1 in the IFN-I Jak-STAT signaling but also acts as a ubiquitin E3 ligase that targets phosphorylated IRF3 and IRF7 (both also targeted by SOCS3 that recruits the Cul-RBX2 E3 complex) for proteasomal degradation in the nucleus [164][179]. As such, PIAS3 acts as a SUMO E3 ligase that inhibits IRF1 transcriptional activity through sumoylation in addition to its ability to inhibit STAT3 [165][180]. TRIM14 negatively regulates IFN-I signaling in mouse macrophage in response to Mycobacterium tuberculosis infection by serving as a scaffold that bridges TBK1-STAT3 interaction promoting STAT3 S727 phosphorylation, consequently inducing SOCS3 expression that inhibits IFN-I signaling by targeting phosphorylated IRF3 and IRF7 as well as TBK1 for proteasomal degradation [83]. The nuclear protein TRIM19/PML promotes ISGF3-mediated gene expression by facilitating STAT1 gene transcription and STAT2 protein stabilization, as well as the accumulation of both activated STAT1 and -2 to chromosome [90].
IKKε is not only responsible for the activation of IRF7 and -3 but also plays a role in balancing IFN-I and IFN-II Jak-STAT signaling pathways in immune responses [166][181]. TRIM6 catalyzes free chains of K48, which promotes IKKε oligomerization and activation to facilitate STAT1 S708 phosphorylation and IFN-I signaling [65]. However, TRIM24 can inhibit retinoic acid-induced STAT1 transcription by interacting with the transcription factor RARα on the STAT1 gene promoter [118].
IFN-Is establish an antiviral state in both virus-infected cells and uninfected bystander cells, by inducing the expression of over 300 ISGs (IFN-stimulated genes) [6]. Many components of the PRR signaling pathways, such as RIG-I, cGAS, STING, IRF1, and IRF7 belong to ISGs. In addition to these components, many other ISGs, including some TRIMs themselves, are also directly regulated by TRIMs. For example, TRIM11 promotes TRIM5 turnover dependently on its RING domain [78]. Ubiquitination-like modifications, such as sumoylation and ISGylation, are involved in IFN-I-mediated defense mechanisms [30][34][167][168][169][170][30,34,182,183,184,185]. TRIM19/PML mediates global sumoylation [92], and TRIM25 functions as an ISG15 E3 ligase that mediates ISGylation [124]. Further, TRIM25 has been recently reported to be required for the stability of several ISG products [171][186]. The zinc-finger antiviral protein ZAP, as an ISG, is activated by TRIM25-mediated ubiquitination to inhibit viral genome translation [125]. The tumor suppressor p53 is also an ISG inducible by IFN-Is [172][187]. TRIM24 promotes p53 ubiquitination and degradation and, in turn, is inducible by p53 [119]. ATM phosphorylates TRIM24 at S768 and promotes its degradation, stabilizing p53 [173][188]. Numerous TRIMs, in addition to TRIM24, regulate p53 activity and stability in direct or indirect manners [174][189].