The serine/threonine kinase, GSK-3, is a promising drug discovery target for treating multiple pathological disorders. Most GSK-3 inhibitors that were developed function as ATP competitive inhibitors, with typical limitations in specificity, safety and drug-induced resistance. In contrast, substrate competitive inhibitors (SCIs), are considered highly selective, and more suitable for clinical practice. The development of SCIs has been largely neglected in the past because the ambiguous, undefined nature of the substrate-binding site makes them difficult to design. In this study, we used our previously described structural models of GSK-3 bound to SCI peptides, to design a pharmacophore model and to virtually screen the “drug-like” Zinc database (~6.3 million compounds). We identified leading hits that interact with critical binding elements in the GSK-3 substrate binding site and are chemically distinct from known GSK-3 inhibitors. Accordingly, novel GSK-3 SCI compounds were designed and synthesized with IC50 values of~1–4 μM. Biological activity of the SCI compound was confirmed in cells and in primary neurons that showed increased β-catenin levels and reduced tau phosphorylation in response to compound treatment. We have generated a new type of small molecule GSK-3 inhibitors and propose to use this strategy to further develop SCIs for other protein kinases.
The serine/threonine kinase, GSK-3, is a promising drug discovery target for treating multiple pathological disorders. Most GSK-3 inhibitors that were developed function as ATP competitive inhibitors, with typical limitations in specificity, safety and drug-induced resistance. In contrast, substrate competitive inhibitors (SCIs), are considered highly selective, and more suitable for clinical practice. The development of SCIs has been largely neglected in the past because the ambiguous, undefined nature of the substrate-binding site makes them difficult to design. In this study, we used our previously described structural models of GSK-3 bound to SCI peptides, to design a pharmacophore model and to virtually screen the “drug-like” Zinc database (~6.3 million compounds). We identified leading hits that interact with critical binding elements in the GSK-3 substrate binding site and are chemically distinct from known GSK-3 inhibitors. Accordingly, novel GSK-3 SCI compounds were designed and synthesized with IC50 values of~1–4 μM. Biological activity of the SCI compound was confirmed in cells and in primary neurons that showed increased β-catenin levels and reduced tau phosphorylation in response to compound treatment. We have generated a new type of small molecule GSK-3 inhibitors and propose to use this strategy to further develop SCIs for other protein kinases.
1. Introduction
Protein kinases (PK) are important regulators of many biological processes, and represent an important class of targets for a diversity of human diseases and pathologies [1][2][3]. Most protein kinase inhibitors developed to date are small molecules that compete with the ATP binding of the kinase. This type of inhibitor, although powerful, often has limited specificity [4][5] because the ATP binding site is highly conserved among protein kinases [2][3]. Indeed, the vast majority of these inhibitors interact and cross-react with multiple members of the PK family [4][5][6], furthermore, they tend to induce drug resistance due to the formation of point mutations at the ATP binding site [6][7][8]. Thus, a different type of inhibitors that do not target the ATP binding site may constitute reliable PKs inhibitors for clinical use.
Protein kinases (PK) are important regulators of many biological processes, and represent an important class of targets for a diversity of human diseases and pathologies [1,2,3]. Most protein kinase inhibitors developed to date are small molecules that compete with the ATP binding of the kinase. This type of inhibitor, although powerful, often has limited specificity [4,5] because the ATP binding site is highly conserved among protein kinases [2,3]. Indeed, the vast majority of these inhibitors interact and cross-react with multiple members of the PK family [4,5,6], furthermore, they tend to induce drug resistance due to the formation of point mutations at the ATP binding site [6,7,8]. Thus, a different type of inhibitors that do not target the ATP binding site may constitute reliable PKs inhibitors for clinical use.
A different class of PK inhibitors, although not extensively studied, represents the substrate competitive inhibitors (SCIs). SCIs interact with the less conserved (and consequently, more specific) substrate-binding cavity of the kinase [2][3] and hold great promise as new therapeutics because they are highly selective, considered safe, and are less prone to drug-induced resistance [9][10][11]. However, SCI development has been somewhat neglected in the past, particularly SCIs in the form of small molecules. Clearly, SCIs were considered difficult to design due to the ambiguous, non-well-defined substrate binding site which is usually large and shallow. Thus, the search and development of SCIs is a challenging task.
A different class of PK inhibitors, although not extensively studied, represents the substrate competitive inhibitors (SCIs). SCIs interact with the less conserved (and consequently, more specific) substrate-binding cavity of the kinase [2,3] and hold great promise as new therapeutics because they are highly selective, considered safe, and are less prone to drug-induced resistance [9,10,11]. However, SCI development has been somewhat neglected in the past, particularly SCIs in the form of small molecules. Clearly, SCIs were considered difficult to design due to the ambiguous, non-well-defined substrate binding site which is usually large and shallow. Thus, the search and development of SCIs is a challenging task.
Target-based docking screens for novel ligands have been largely used over the last decades and became principal tools in drug discovery [12][13][14][15]. The prior knowledge of protein–ligand intermolecular interactions is indeed a key for the identification of reliable hits that will later serve as successful starting points for drug design. However, the scarce number of available structures of PKs bound to their substrates discouraged perusing in silico-based searches for PKs-SCI. Indeed, to the best of our knowledge, computational approaches have not been practiced for the identification of small molecules SCIs.
Target-based docking screens for novel ligands have been largely used over the last decades and became principal tools in drug discovery [12,13,14,15]. The prior knowledge of protein–ligand intermolecular interactions is indeed a key for the identification of reliable hits that will later serve as successful starting points for drug design. However, the scarce number of available structures of PKs bound to their substrates discouraged perusing in silico-based searches for PKs-SCI. Indeed, to the best of our knowledge, computational approaches have not been practiced for the identification of small molecules SCIs.
The serine-threonine kinase, GSK-3, is a validated target for drug discovery in treating several pathologies including diabetes, neuronal development, neurodegenerative diseases and psychiatric disorders [16][17][18][19][20][21]. In humans, GSK-3 is expressed as two isozymes, GSK-3α and GSK-3β, which are encoded by two genes and share high homology in their catalytic domains [22]). The mechanisms by which GSK-3 is thought to contribute to pathogenesis are diverse. These include phosphorylation of the microtubule-associated protein tau [23][24], destabilization of the Wnt signaling component β-catenin [25][26], regulation of multiple transcript factors such as NF-κB [27][28], activation of pro-inflammatory factors [29], and impairment of cellular clearance pathways [30][31]. Inhibition of GSK-3 was considered a promising therapeutic approach, however, GSK-3 ATP competitive inhibitors failed in the pre-clinical phase due to toxicity and side effects.
The serine-threonine kinase, GSK-3, is a validated target for drug discovery in treating several pathologies including diabetes, neuronal development, neurodegenerative diseases and psychiatric disorders [16,17,18,19,20,21]. In humans, GSK-3 is expressed as two isozymes, GSK-3α and GSK-3β, which are encoded by two genes and share high homology in their catalytic domains [22]). The mechanisms by which GSK-3 is thought to contribute to pathogenesis are diverse. These include phosphorylation of the microtubule-associated protein tau [23,24], destabilization of the Wnt signaling component β-catenin [25,26], regulation of multiple transcript factors such as NF-κB [27,28], activation of pro-inflammatory factors [29], and impairment of cellular clearance pathways [30,31]. Inhibition of GSK-3 was considered a promising therapeutic approach, however, GSK-3 ATP competitive inhibitors failed in the pre-clinical phase due to toxicity and side effects.
In previous works, we developed a series of short phosphorylated SCI peptides for GSK-3. The SCI peptides were derived from the unique substrate-recognition motif of GSK-3, S
1
XXXS
2
(p) (where S
1
is the GSK-3-phosphorylation site and S
2 is the phosphorylated priming site) [32][33] and are patterned after the GSK-3 substrate heat shock factor-1 (HSF-1) [34]). The combined computational and biochemical analysis identified the critical sites important for the GSK-3 SCI peptides interactions with the GSK-3 substrate-binding site [33][35]. GSK-3 binds the “primed” phosphorylated peptide inhibitor (that mimics the primed phosphorylated substrate) through its “phosphate-binding pocket” (Arg 96, Arg 180, and Lys 205) [36][37]. In addition, the substrate binds to a segment bordered by Gln 89-Asn 95, termed the “89–95” loop in which Phe 93 is the most critical residue for binding [33][35]. In some cases, the SCI peptides interact with a “hydrophobic patch” (Val 214, Ile 217, and Tyr 216) located in proximity to the enzyme’s phosphate-binding pocket [33]). The therapeutic potential of our SCI peptides was demonstrated in several cellular and disease mouse models including Parkinson’s, Alzheimer’s disease, multiple sclerosis, depressive behavior, and Fragile X syndrome [31][32][38][39][40][41][42]. Here we aimed to develop small molecule SCIs for GSK-3. Based on our structural GSK-3-SCI peptide-binding model we discovered and designed novel GSK-3 SCI molecules and confirmed their biological activity in vitro and in cellular systems.
is the phosphorylated priming site) [32,33] and are patterned after the GSK-3 substrate heat shock factor-1 (HSF-1) [34]). The combined computational and biochemical analysis identified the critical sites important for the GSK-3 SCI peptides interactions with the GSK-3 substrate-binding site [33,35]. GSK-3 binds the “primed” phosphorylated peptide inhibitor (that mimics the primed phosphorylated substrate) through its “phosphate-binding pocket” (Arg 96, Arg 180, and Lys 205) [36,37]. In addition, the substrate binds to a segment bordered by Gln 89-Asn 95, termed the “89–95” loop in which Phe 93 is the most critical residue for binding [33,35]. In some cases, the SCI peptides interact with a “hydrophobic patch” (Val 214, Ile 217, and Tyr 216) located in proximity to the enzyme’s phosphate-binding pocket [33]). The therapeutic potential of our SCI peptides was demonstrated in several cellular and disease mouse models including Parkinson’s, Alzheimer’s disease, multiple sclerosis, depressive behavior, and Fragile X syndrome [31,32,38,39,40,41,42]. Here we aimed to develop small molecule SCIs for GSK-3. Based on our structural GSK-3-SCI peptide-binding model we discovered and designed novel GSK-3 SCI molecules and confirmed their biological activity in vitro and in cellular systems.
2. Results
2.1. The Use of GSK-3/SCI Peptide Binding Model for Pharmacophore-Based Virtual Screening
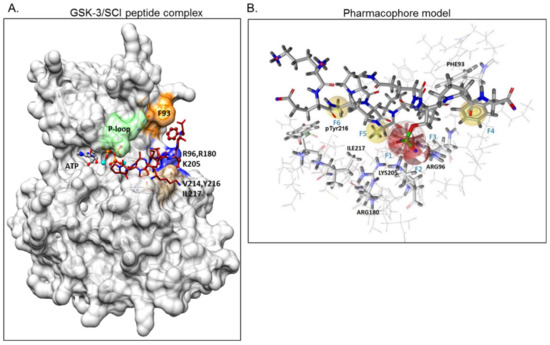
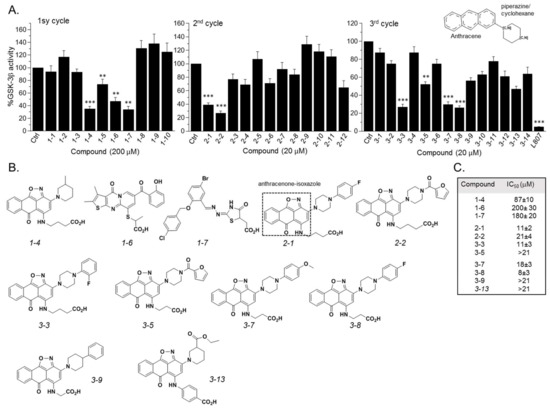
GSK-3β binding with the SCI peptide L803F (KEAPPSPPQS(p)PF) is shown in A. This peptide is an improved version of our “original” L803 peptide inhibitor (KEAPPSPPQS(p)P) [33][43]. The model highlights the important features in substrate/SCI binding into the GSK-3β catalytic groove: the phosphorylated serine interacts with the phosphate-binding pocket of the kinase (Arg 96, Arg 180, and Lys 205) [36][37], prolines and phenylalanine in L803F interact with Phe 93, a part of the “89–95” loop, and a critical residue in substrate binding [33], the N-terminal end of L803F interacts with a “hydrophobic patch” in GSK-3β composed of Tyr 216, Val 214, and Ile 217 (
A. This peptide is an improved version of our “original” L803 peptide inhibitor (KEAPPSPPQS(p)P) [33,43]. The model highlights the important features in substrate/SCI binding into the GSK-3β catalytic groove: the phosphorylated serine interacts with the phosphate-binding pocket of the kinase (Arg 96, Arg 180, and Lys 205) [36,37], prolines and phenylalanine in L803F interact with Phe 93, a part of the “89–95” loop, and a critical residue in substrate binding [33], the N-terminal end of L803F interacts with a “hydrophobic patch” in GSK-3β composed of Tyr 216, Val 214, and Ile 217 (A) [33]. Thus, small molecules that mimic this binding mode should function as potent SCI molecules and we set out to discover such molecules. The similarity to 1-4
, 1-6
, and 1-7
(The hits 241 analogues for 1-4
, 11 analogues for 1-6
, and 21 analogues for 1-7
) were filtered as described above, and 12 available compounds were purchased (Table S2
) and tested (A). Compounds 2-1
and 2-2
(1-4
analogues, B) acted as best GSK-3β inhibitors (A,C). As 1-4
analogues seemed more efficacious than the analogues related to 1-6
or 1-7
, we conducted a third search looking for molecules with the substructure core of 1-4
: anthracene attached to a piperazine/cyclohexane ring (see illustration, A). We reasoned that suitable bioactive analogues could have been overlooked by our initial search, in addition, the fact that ZINC database is updated on a regular basis, and new compounds may become available. This search identified 137 hits, in which 15 were compatible with an interaction with the GSK-3β site, and 14 available compounds were purchased (Table S3
). In vitro GSK-3 kinase assays indicated that most of the hit compounds acted as GSK-3β inhibitors (A). The SCI peptide L807mts [32]) was used as a reference in these assays (A 3rd cycle). Among them, 3-3
, 3-5
, 3-7
, 3-8
, 3-9
and 3-13
demonstrated best performance as GSK-3β inhibitors with IC50
values of ~8–20 μM. (B,C).
Figure 1.
Design of a pharmacophore model based on GSK-3β binding model with substrate competitive inhibitor (SCI) peptide (
A
) Structural model of GSK-3β bound to our previously described SCI peptide L803F. The primed. phosphate S
10
(p) in the peptide interacts with the phosphate-binding pocket (Arg 96, Arg 180, Ly 205, marked blue), Phe
12
, at the C-terminal end of the peptide, interacts with Phe
93
located in the “89–95” loop (orange), Ala
3
in the peptide interacts with pTyr
216
, and Pro
5
in the peptide interacts with Val
214
and Ile
217
, all residues that form the “hydrophobic patch” (beige). The P-loop including Phe
67
is in green, the ATP molecule and Mg
+2
(cyan balls) are shown. (
B
) The pharmacophore model is composed of two hydrogen bond acceptor features (F1, F2: red vectors), one anionic feature (F3: red porcupine shape), and three hydrophobic features (F4, F5, F6: yellow spheres). Excluded volumes were placed in positions that are sterically claimed by the protein environment. GSK-3 interacting residues are marked.
Figure 2.
Discovery of GSK-3 SCI hits. (
A
) In vitro GSK-3β kinase assays were conducted with selected hits identified in the three iterative virtual screening cycles. Structure of compounds is summarized in
Tables S1–S3
. Ctrl represents GSK-3 activity no compounds (100%). Results are mean of three independent experiments ± SEM using one-way ANOVA with Dunnett’s post hoc test. **
p
< 0.01 ***
p
< 0.001. The anthracene core used in the third search cycle is illustrated at the top right panel. The SCI peptide L807mts (L807) was used as a reference in the 3rd cycle assays. (
B
) Chemical structures of selected best hits of each cycle. The anthracenone–isoxazole core is highlighted in compound
2-1
. (
C
) IC
50
values of selected best hits.
2.2. SCI Hits Interact with the GSK-3β Substrate Binding Site and Are Chemically Unique
Docking analysis indeed showed that the identified compounds matched the pharmacophore requirements and shared similar structural features (). Representative docking models of GSK-3β with the best hits 3-7
and 3-8
are also shown in A. Clearly, all discovered compounds shared a common carboxylic acid moiety that interacted with the phosphate-binding pocket (Arg 96, Arg 180, Lys 205) through salt bridges and H-bonds. This novel observation indicated for the first time that a non-phosphorylated group can mimic the phosphorylated substrates of GSK-3. In some cases, the carboxylic acid moiety formed H-bonds with Val 214, which also participates in GSK-3-L803F binding (A) [33]. Most of the compounds contained an anthracenone–isoxazole core that formed π–π stacking and/or cation–π interactions with Phe 93 and Arg 96 respectively. In some instances, N or O at the isoxazol ring formed H-bonds with Arg 96, and the benzoic acid moieties interacted with Lys 85, Asn 95, and Glut-97, all important residues for kinase catalytic activity: Lys 85 and Glut 97 are highly conserved residues that facilitate interactions with ATP, and Asn 95 participate in substrate binding [33][44][45]. Take, together, our discovered compounds captured the binding mode of the GSK-3-SCI peptide complex.
A) [33]. Most of the compounds contained an anthracenone–isoxazole core that formed π–π stacking and/or cation–π interactions with Phe 93 and Arg 96 respectively. In some instances, N or O at the isoxazol ring formed H-bonds with Arg 96, and the benzoic acid moieties interacted with Lys 85, Asn 95, and Glut-97, all important residues for kinase catalytic activity: Lys 85 and Glut 97 are highly conserved residues that facilitate interactions with ATP, and Asn 95 participate in substrate binding [33,44,45]. Take, together, our discovered compounds captured the binding mode of the GSK-3-SCI peptide complex.
Figure 3.
GSK-3 SCI compounds interact with the kinase substrate binding site and are chemically unique. (
A
) Docking models of compounds
3-7
and
3-8
with GSK-3β. Key interacting GSK-3β residues are highlighted, and detailed interactions are summarized in
. The carboxylic acid moiety form salt bridges and H-bonding with the GSK-3-phosphate-binding pocket, Arg 96, Arg 180, Lys 205, and with Val 214. The anthracenone–isoxazole core forms π–π stacking interactions with Phe 93. Interactions were analyzed by the Maestro software. (
B
) In vitro kinase assays were performed with GSK-3β (WT) or with the F93A mutant in the presence of indicated compounds, L803F (20 μM each), and SB216763 (1 μM). Results are mean of three independent experiments ± SEM analyzed by one way ANOVA with Dunnett’s multiple comparisons. **
p
< 0.01 with inhibitor vs no inhibitor (
C
) Principal component analysis (PCA) analysis of all compounds discovered through the three search cycles together with representative GSK-3-ATP competitive inhibitors (listed in
Table S4
). The first and second PCs accounted for 81.8% and 7.7% of the original variance and are shown at the
X-
and
Y
-axis respectively. Black circles represent the ATP competitive inhibitors, colored circles represent compounds from cycle 1 (green), 2 (red) and 3 (blue). Circles with yellow dots represent compounds
2-1
,
3-7
and
3-8
.
Table 1.
GSK-3 interactions with selected compounds. The number of interactions (*), type of interaction, and atom/substructure involved are listed for each GSK-3 residue. Green, H-bond, blue πι–π stacking, black, cation–π, pink, salt bridge.
| Name |
GSK-3 Interacting Residues |
| Arg 96 |
Arg 180 |
Lys 205 |
Phe 93 |
Val 214 |
Others |
| 1-4 |
* Carbonyl of anthracenone
* O of CO2H |
** 2O of CO2H |
** O of CO2H |
|
|
* Gly 202 with amine
at the CO2H chain |
| 1-6 |
** thiophene ring
Carbonyl of anthracenone,
* O of CO2H |
*O of CO2H |
*O of CO2H |
|
|
* Glut 97 with phenol hydroxyl and ketone,
* Asn 95 with phenol hydroxyl |
| 1-7 |
|
**** Carbonyl at thiophene ring
2O of CO2H
O of CO2H |
* O of CO2H |
* Bromophenyl
ring |
* O of CO2H |
|
| 2-1 |
*** 2O of CO2H
N of isoxazole |
*** 2O of CO2H
O of CO2H |
* O of CO2H |
*** Isoxazole and aromatic rings of anthracenone |
* O of CO2H |
|
| 2-2 |
*** Carbonyl at the anthracenone
benzoic acid
O of CO2H |
** O of CO2H
benzoic acid |
** 2O of CO2H |
|
|
* Gly 202 with amine attached to benzoic acid
* Lys 85 with O of ether group |
| 3-7 |
*** O of CO2H |
** 2O of CO2H
* O of CO2H |
* O of CO2H
* O of CO2H |
** Isoxazole and aromatic rings of anthracenone |
* O of CO2H |
|
| 3-8 |
** 2O of CO2H |
** 2O of CO2H
* O of CO2H |
** O of CO2H |
** Isoxazole and aromatic rings of anthracenone |
* O of CO2H |
To further confirm that compounds interact with the GSK-3β substrate binding site, we performed in vitro kinase assays with the GSK-3β-F93A mutant in which Phe 93 was replaced by alanine. This mutant is active but does not bind substrates or SCI peptides efficiently [33]. Indeed, like the SCI peptide L803F, F93A was barely inhibited by the SCI compounds (B). This, in contrast, to SB216763, an established GSK-3 ATP competitive inhibitor [46] that inhibited both wild-type (WT) and the F93A mutant (B).
Finally, we conducted a principal component analysis (PCA) to determine whether the SCI compounds are chemically distinct from other known GSK-3 inhibitors. This was performed to confirm that the discovered compounds are unique and worthy of further development. Briefly, PCA projects a dataset originally described in a high-dimensional space into a 2-dimensional space while keeping, as much as possible, the original distribution of the data points (i.e., the distances between them). The analysis was performed on all SCI compounds (Tables S1–S3
) together with representative ATP competitive GSK-3 inhibitors including SB-216763, CHIR98014, AR-A014418, VP2.51, Bio6, Kenpaullone, and 1-Azakenpaullone (listed in Table S4
). The analysis clearly showed that the SCI compounds occupy a chemical space distinct from that occupied by the ATP competitive inhibitors (C). The PCA analysis also revealed the successive focusing toward the active compounds region obtained through the three search cycles. These results encouraged further development of the discovered hits.
2.3. Design and Synthesis of Novel GSK-3 SCIs
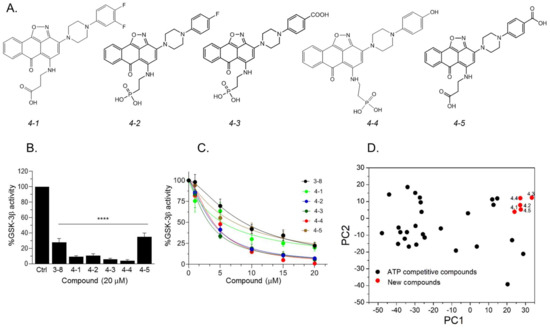
Our results so far indicated that the discovered hits can represent a new class of GSK-3 inhibitors that act as SCIs. Based on the leading compounds 3-7
and 3-8
, we designed and synthesized new SCI molecules. We first attempted to replace the carboxylic acid moiety (CH2
CO2
H) with a phosphonate group (CH2
PO3
H2) to better mimics the “native” phosphorylation in GSK-3 substrates (GSK-3 substrate are pre-phosphorylated) [32][33]. In addition, we assumed that substituting the fluorine atom at the aromatic ring or other electron-withdrawing group such as CO
) to better mimics the “native” phosphorylation in GSK-3 substrates (GSK-3 substrate are pre-phosphorylated) [32,33]. In addition, we assumed that substituting the fluorine atom at the aromatic ring or other electron-withdrawing group such as CO2
H, or, OH, or an additional fluorine atom to the ring, will facilitate additional contacts with the kinase. The newly synthesized compounds termed 4-1
, 4-2
, 4-3
, 4-4
, and 4-5
are presented in A. In vitro kinase assays confirmed that the new compounds inhibited GSK-3β and were indeed better inhibitors as compared to 3
-8 (B,C). Collectively, compounds 4-3
and 4-4
acted as best inhibitors showing IC50
values of ~1–4 μM. PCA analysis further confirmed that the new compounds are chemically distinct from other GSK-3 inhibitors (D).
Figure 4.
Newly designed GSK-3 SCI molecules. (
A
) Structures of new molecules
4-1
to
4-5
. (
B
) In vitro GSK-3β kinase assays were conducted with new compounds and
3-8
(20 μM each). Results show the percentage of GSK-3β activity without inhibitor (100%) and represent the mean of three independent experiments ± SEM analyzed by one way ANOVA with Dunnett’s multiple comparisons. ****
p
< 0.0001. (
C
) Dose–response curves of GSK-3β inhibition of new compounds and as compared to
3-8
. Results are mean of three independent experiments ± SEM. For
4-3
and
4-4
*
p
< 0.05 for all concentrations, for the rest of the molecules, *
p
< 0.05 at concentrations ≥ 5 μM as determined by one way ANOVA with Dunnett’s multiple comparisons, new compounds vs.
3-8
. (
D
) PCA analysis of new compounds
4-1
to
4-5
together with GSK-3-ATP competitive inhibitors (listed in
Table S4
). The first and second PCs accounted for 81.8% and 7.7% of the original variance and are shown at the
X
- and
Y
-axis respectively. Black circles represent the ATP competitive inhibitors, red circles represent new compounds.
Representative docking models of GSK-3β bound to 4-2
, 4-3
and 4-4
are shown in , and detailed interactions are summarized in . The new compounds showed similar docking poses as those produced with 3-8
. As expected, the PO3
H2
moiety interacted with the phosphate-binding pocket, and the anthracenone–isoxazole core formed π–π stacking interactions with Phe 93. Compounds 4-3
and 4-4
formed additional interactions with N or O of the isoxazole ring, and 4-4
formed cation–π interactions with Arg 96. In addition, 4-3
and 4-4
extended their interactions with Phe 67, Lys 85, or, Ser 66 through their CO2H or OH moieties respectively. These residues play a role in GSK-3-substrate binding [35][45]. Thus, the additional interactions performed with
H or OH moieties respectively. These residues play a role in GSK-3-substrate binding [35,45]. Thus, the additional interactions performed with