Immune checkpoint blockade targeting PD-1/PD-L1 has a promising therapeutic efficacy in different tumors, but a significant percentage of patients cannot benefit from this therapy due to primary and acquired resistance during treatment. Key mechanisms about modulating the immunosuppressive tumor microenvironment, such as depletion of Tregs, IDO, or MDSCs, interfering suppressive cytokines and inhibiting alternative immune checkpoints, may enhance the therapeutic efficacy of the PD-1/PD-L1 blockade.
- PD-L1
- resistance
- immune checkpoints
1. Introduction
T-cell activation and proliferation induced by antigens is regulated by expression of both co-stimulatory and co-inhibitory receptors and their ligands [1]. Inhibitory pathways in the immune system can prevent autoimmunity through maintaining self-tolerance and regulating immunity [2]. While in tumors inhibitory pathways known as “checkpoints” can evade immune surveillance. Programmed cell death -1(PD-1) interacting with its corresponding ligand PD-L1 leads to immune suppression via preventing the T-cell activation in the tumor [3]. PD-1 is expressed on activated CD8
+
T-cells as well as B cells and natural killer cells, and inhibits T-cell receptor (TCR) signaling and CD28 co-stimulation under chronic antigen exposure. As ligands of PD-1, PD-L2 is primarily expressed on antigen-presenting cells (APC) while PD-L1 is expressed on various types of cells including tumor cells and immune cells. Evidence of PD-L1 expression increase and spontaneous immune resistance is proved in several types of human cancers [4]. Besides, predictive and prognostic value of PD-L1 immunohistochemical expression has been reported in certain cancers. Moreover, PD-L1 as an inhibitory factor is also involved in other signaling pathways underlying mechanisms in resistance to tyrosine kinase inhibitors (TKIs).
Immunotherapy identified as the most promising approach in cancer treatment compared with chemotherapy and targeted therapy, immune checkpoint inhibitors have reported higher rates of response, remission, and better overall survival rates in a variety of tumors [5]. Immunotherapy has received the US Food and Drug Administration (FDA) approval for 57 indications in 17 solid tumors in less than 10 years, while over 80% are PD-1/PD-L1-targeted antibodies. Beneficial function of the PD-1/PD-L1 axis blockade is confirmed in treating many different types of cancers such as non-small cell lung cancer (NSCLC), melanoma and bladder carcinoma [6][7][6,7]. So far, six immune checkpoint inhibitors targeting PD-1/PD-L1 have been approved by the FDA for the first and second line of patients with non-small cell lung cancer including monoclonal antibodies (mAb) pembrolizumab, nivolumab and cemiplimab targeting PD-1 and mAb atezolizumab, avelumab and durvalumab targeting PD-L1. However, limited efficacy has been reported in PD-1/PD-L1 blockade therapy which rarely exceeds 40% in most cancer types and a large number of patients show partial responsiveness [8][9][8,9]. Even if there is a consistent rate of initial responses, the majority of patients develop therapeutic resistance and disease progression [10][11][10,11].
2. Key Mechanisms Underlying Resistance to PD-1/PD-L1 Blockade
PD-1/PD-L1 blockade therapy has been approved as a significantly helpful treatment in certain cancers, a problem of its limited efficacy has occurred and the targeting solution is urgently discussed and provided. Focusing on PD-L1, we described key mechanisms underlying resistance to PD-1/PD-L1. Surprisingly, abnormally upregulated PD-L1 expression and a lack of PD-L1 can both lead to inefficacy of PD-1/PD-L1 inhibitors (Figure 1).
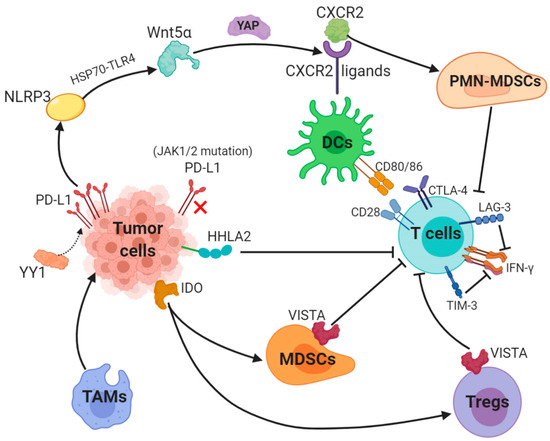
Figure 1. Key mechanisms underlying resistance to PD-L1 (1). The transcription factor Yin Yang 1 (YY1)-induced upregulation of PD-L1 expression triggers NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome to promote tumor Wnt5α expression via HSP70-TLR4 signaling, and non-canonical WNT ligands activate the YAP pathway to induce chemokine (C-X-C motif) receptor 2 (CXCR2) ligands, while granulocytic subset of myeloid-derived suppressor cells (PMN-MDSCs) relied on CXCR2 to suppress T-cell function. (2) Loss-of-function mutations in JAK1/2 leads to the paucity of PD-L1 expression. (3) Tumor-suppressing microenvironment. Tumor-associated macrophages (TAMs) promote tumor progression, while Indole 2,3-dioxygenase (IDO) generated by tumors enhances Tregs and MDSCs activity, which suppress immunity. (4) Activation of alternative immune checkpoints. T-cell immunoglobulin mucin 3 (TIM-3) and Lymphocyte activation gene-3 (LAG-3) produced by T-cells impair generation of IFN-γ, which activates T-cells. CTLA-4 demonstrates a higher affinity and avidity in conjunction with CD80 and CD86 than CD28 to antagonize costimulation. VISTA is found to be related to MDSC mainly derived CD33 expression and HHLA2 decreases T-cell proliferation.
2.1. Aberrant PD-L1 Expression
PD-L1 is generally regulated by tumor cells in two ways: the first is innate immune resistance in which constitutive oncogenic signaling is correlated with PD-L1 expression, the second is an adaptive immune resistance through which IFN-γ produced by TILs induces PD-L1 expression.K-ras mutation as a common oncogenic driver in the lung adenocarcinoma (LUAD) and upregulates PD-L1 through p-ERK instead of p-AKT signaling [12]. Different subgroups of KRAS-mutant LUAD are dependent on STK11/LKB1 or TP53 mutations, and alterations of the former has been confirmed as a major factor that leads to primary resistance to PD-1 blockade [13]. Besides, EGFR-mutant or ALK-rearranged patients had a PD-L1 tumor proportion score of ≥50% and turned out not to respond to PD-1/PD-L1 inhibitors [14].The transcription factor Yin Yang 1 (YY1); a major regulator reported participating in various pathways, is involved in cell growth, survival and metastasis. YY1 upregulates PD-L1 expression on tumor cells via signaling pathways, including p53, STAT3, NF-κB and PI3K/AKT/mTOR [15]. PD-L1v242 and PD-L1v229, two secreted PD-L1 C-terminal splicing variants, could capture the aPD-L1 antibody and function as a “decoy” to prevent antibodies from binding to PD-L1 [16]. Besides, a tumor-intrinsic signaling pathway involved with NLRP3 inflammasome in response to upregulated expression of PD-L1 was found to drive adaptive resistance to anti-PD-1 antibody immunotherapy [17]. NLRP3 inflammasome triggered by PD-L1 induces tumor Wnt5α expression via HSP70-TLR4 signaling, while non-canonical WNT ligands promote production of CXCR2 ligands through the activated YAP pathway [18][19]. CXCR2-relied migration and recruitment of a granulocytic subset of MDSCs (PMN-MDSCs) play a role in suppressing CD8
2.1. Aberrant PD-L1 ExpressionPD-L1 is generally regulated by tumor cells in two ways: the first is innate immune resistance in which constitutive oncogenic signaling is correlated with PD-L1 expression, the second is an adaptive immune resistance through which IFN-γ produced by TILs induces PD-L1 expression.K-ras mutation as a common oncogenic driver in the lung adenocarcinoma (LUAD) and upregulates PD-L1 through p-ERK instead of p-AKT signaling [53]. Different subgroups of KRAS-mutant LUAD are dependent on STK11/LKB1 or TP53 mutations, and alterations of the former has been confirmed as a major factor that leads to primary resistance to PD-1 blockade [54]. Besides, EGFR-mutant or ALK-rearranged patients had a PD-L1 tumor proportion score of ≥50% and turned out not to respond to PD-1/PD-L1 inhibitors [55].The transcription factor Yin Yang 1 (YY1); a major regulator reported participating in various pathways, is involved in cell growth, survival and metastasis. YY1 upregulates PD-L1 expression on tumor cells via signaling pathways, including p53, STAT3, NF-κB and PI3K/AKT/mTOR [56]. PD-L1v242 and PD-L1v229, two secreted PD-L1 C-terminal splicing variants, could capture the aPD-L1 antibody and function as a “decoy” to prevent antibodies from binding to PD-L1 [57].Besides, a tumor-intrinsic signaling pathway involved with NLRP3 inflammasome in response to upregulated expression of PD-L1 was found to drive adaptive resistance to anti-PD-1 antibody immunotherapy [58]. NLRP3 inflammasome triggered by PD-L1 induces tumor Wnt5α expression via HSP70-TLR4 signaling, while non-canonical WNT ligands promote production of CXCR2 ligands through the activated YAP pathway [59,60]. CXCR2-relied migration and recruitment of a granulocytic subset of MDSCs (PMN-MDSCs) play a role in suppressing CD8+ T-cell infiltration and function, therefore leading to adaptive resistance [20][21]. Previous study showed that tumors can be divided into four categories according to positive/negative tumor PD-L1 expression and presence/absence of TILs. For instance, patients with PD-L1 positive and TILs indicate adaptive immune resistance and those with PD-L1 negative and without TILs show immune ignorance [22]. Among these four types, type I with PD-L1 positive and TILs is the most likely to respond to PD-1/PD-L1 blockade therapy, whilst other types may show unresponsiveness to this monotherapy [23].
T-cell infiltration and function, therefore leading to adaptive resistance [61,62].Previous study showed that tumors can be divided into four categories according to positive/negative tumor PD-L1 expression and presence/absence of TILs. For instance, patients with PD-L1 positive and TILs indicate adaptive immune resistance and those with PD-L1 negative and without TILs show immune ignorance [63]. Among these four types, type I with PD-L1 positive and TILs is the most likely to respond to PD-1/PD-L1 blockade therapy, whilst other types may show unresponsiveness to this monotherapy [64].2.2. Paucity of PD-L1 Expression
The interaction between PD-L1 and its receptor PD-1 leads to immune escape and inhibits T-cell function and blockade of PD-L1 and PD-1 enhances the antitumor immunity in several cancers. However, the expression of PD-L1 or PD-1 is a prerequisite for the therapeutic efficacy. Evidence of the relation of rare PD-L1 expression and poorer responses to PD-1 blockade has been proved in prostate cancer [24]. DNA hypomethylating agent upregulate PD-L1 gene expression [25]. Anti-PD-1 therapy curbs the expression of PD-L1 through either eliminating the tumor cells that overexpress PD-L1 and possess a hypomethylated PD-L1 promoter or switching off the PD-L1 expression through epigenetic modulation, therefore leading to resistance [26]. Loss-of-function mutations in JAK1/2 can lead to primary resistance to anti-PD-1 therapy due to the inability to respond to IFN-γ for a lack of PD-L1 expressions [27]. Despite the effect of aberrant PD-L1 expression, an abnormal process from antigen expression to T-cell activation can result in resistance to PD-1/PD-L1 inhibitors. Moreover, a recent study demonstrated that PD-L1 expression is enhanced via nicotinamide adenine dinucleotide (NAD
2.2. Paucity of PD-L1 ExpressionThe interaction between PD-L1 and its receptor PD-1 leads to immune escape and inhibits T-cell function and blockade of PD-L1 and PD-1 enhances the antitumor immunity in several cancers. However, the expression of PD-L1 or PD-1 is a prerequisite for the therapeutic efficacy. Evidence of the relation of rare PD-L1 expression and poorer responses to PD-1 blockade has been proved in prostate cancer [65]. DNA hypomethylating agent upregulate PD-L1 gene expression [66]. Anti-PD-1 therapy curbs the expression of PD-L1 through either eliminating the tumor cells that overexpress PD-L1 and possess a hypomethylated PD-L1 promoter or switching off the PD-L1 expression through epigenetic modulation, therefore leading to resistance [67]. Loss-of-function mutations in JAK1/2 can lead to primary resistance to anti-PD-1 therapy due to the inability to respond to IFN-γ for a lack of PD-L1 expressions [68]. Despite the effect of aberrant PD-L1 expression, an abnormal process from antigen expression to T-cell activation can result in resistance to PD-1/PD-L1 inhibitors. Moreover, a recent study demonstrated that PD-L1 expression is enhanced via nicotinamide adenine dinucleotide (NAD+) metabolism, in which nicotinamide phosphoribosyltransferase (NAMPT) functions as the rate-limiting enzyme [28]. NAMPT increases PD-L1 expression induced by IFN-γ and leads to immune escape in tumors with the help of CD8
) metabolism, in which nicotinamide phosphoribosyltransferase (NAMPT) functions as the rate-limiting enzyme [69]. NAMPT increases PD-L1 expression induced by IFN-γ and leads to immune escape in tumors with the help of CD8+
T-cells. Thus NAD+ metabolism is a promising strategy for resistance to anti-PD-L1 therapy [28].
metabolism is a promising strategy for resistance to anti-PD-L1 therapy [69].2.3. Aberrant Antigen Expression, Presentation and Recognition
Tumors with a higher tumor mutation burden (TMB) are likely to have more neoantigens, which can be recognized by the immune system as “non-self” in response to checkpoint inhibition. In Naiyer’s study, the result of the treatment of PD-1 targeting antibody pembrolizumab in NSCLC described that a higher burden of nonsynonymous tumors is correlated with a better response and PFS [29]. Besides, strong immunogenicity and extensive expression of immune checkpoint ligands make the microsatellite instability subtype more susceptible to immunotherapeutic methods, for example, with anti-PD-L1 and anti-cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) antibodies [30]. Tumors with defective mismatch repair possess more DNA mutations and show an improved responsiveness to anti-PD-1 therapy [31]. In short, a low mutational burden, microsatellite stability and efficient DNA repair mechanisms are involved in innate resistance to immune-checkpoint blockade therapy. Moreover, evolution of neoantigen loss can produce an acquired resistance [32]. A study also demonstrates that deficiency of heterogeneity in HLA genes is observed in cancer development, a high level of HLA loss results in acquired resistance during immunotherapy [33]. Resistance to immune checkpoint blockades also involves impaired DC maturation, which is an essential process in T-cell activation, through it is displayed in various co-stimulatory factors expression including MHC class I/II, CD80, CD86 and CD40 [34]. IL37b decreases CD80 and CD86 expression through the ERK/S6K/NF-κB axis and suppresses DC maturation [35]. A transcription factor STAT3 that facilitates tumor growth and metastasis leads to the induction of other immunosuppressive factors that possess a suppressive function on DC maturation, including IL10, Tregs and TGF-β [36][37][38][39]. Despite inducing PD-L1, IFNs have been reported to (re-)activate T-cells to control the tumor development via advancing DC cross-priming [39][40][41]. It is well-known that CTLs recognize MHC class I-presented peptide antigens on the surface of tumor cells. Heterozygous mutations, deletions or deficiency in β-2-microglobulin (β2M); a crucial factor in MHC class I antigen presentation, generally reduces antigen recognition by antitumor CD8
2.3. Aberrant Antigen Expression, Presentation and RecognitionTumors with a higher tumor mutation burden (TMB) are likely to have more neoantigens, which can be recognized by the immune system as “non-self” in response to checkpoint inhibition. In Naiyer’s study, the result of the treatment of PD-1 targeting antibody pembrolizumab in NSCLC described that a higher burden of nonsynonymous tumors is correlated with a better response and PFS [70]. Besides, strong immunogenicity and extensive expression of immune checkpoint ligands make the microsatellite instability subtype more susceptible to immunotherapeutic methods, for example, with anti-PD-L1 and anti-cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) antibodies [71]. Tumors with defective mismatch repair possess more DNA mutations and show an improved responsiveness to anti-PD-1 therapy [72]. In short, a low mutational burden, microsatellite stability and efficient DNA repair mechanisms are involved in innate resistance to immune-checkpoint blockade therapy. Moreover, evolution of neoantigen loss can produce an acquired resistance [73]. A study also demonstrates that deficiency of heterogeneity in HLA genes is observed in cancer development, a high level of HLA loss results in acquired resistance during immunotherapy [74].Resistance to immune checkpoint blockades also involves impaired DC maturation, which is an essential process in T-cell activation, through it is displayed in various co-stimulatory factors expression including MHC class I/II, CD80, CD86 and CD40 [75]. IL37b decreases CD80 and CD86 expression through the ERK/S6K/NF-κB axis and suppresses DC maturation [76]. A transcription factor STAT3 that facilitates tumor growth and metastasis leads to the induction of other immunosuppressive factors that possess a suppressive function on DC maturation, including IL10, Tregs and TGF-β [77,78,79,80].Despite inducing PD-L1, IFNs have been reported to (re-)activate T-cells to control the tumor development via advancing DC cross-priming [81,82,83]. It is well-known that CTLs recognize MHC class I-presented peptide antigens on the surface of tumor cells. Heterozygous mutations, deletions or deficiency in β-2-microglobulin (β2M); a crucial factor in MHC class I antigen presentation, generally reduces antigen recognition by antitumor CD8+ T-cells and mutation of β2M gene leads to resistance to anti-PD-1 therapy [42][43]. IFN-γ can induce tumor cells to express MHC class I molecules, significantly promoting CTL differentiation and enhancing apoptosis. Mutations or loss of IFN-γ pathway-related proteins on tumor cells (such as STATs, IFN-γ receptor chain JAK1 and JAK2) can cause escape from immune recognition and resistance to immune checkpoint inhibitions [27][44].
T-cells and mutation of β2M gene leads to resistance to anti-PD-1 therapy [84,85]. IFN-γ can induce tumor cells to express MHC class I molecules, significantly promoting CTL differentiation and enhancing apoptosis. Mutations or loss of IFN-γ pathway-related proteins on tumor cells (such as STATs, IFN-γ receptor chain JAK1 and JAK2) can cause escape from immune recognition and resistance to immune checkpoint inhibitions [68,86].2.4. Aberrant Immunity of T-Cells
Despite normal antigen expression, presentation, recognition and successfully activated T-cells, resistance to the PD-1/PD-L1 blockade inhibitors may occur owing to the T-cell itself. The aberrant immunity of T-cells include insufficient T lymphocytes infiltration, dysfunction of T-cell and exhausted T-cells.
3.Tumor-Suppressing Microenvironment
Apart from abnormal T-cells and PD-L1 expression, there are some other types of cells and cytokines that benefit tumor development inside the tumor microenvironment, they form the tumor-suppressing microenvironment to play a key role in resistance to the PD-1/PD-L1 blockade.
4. Activation of Alternative Immune Checkpoints
As one of the most prospective approaches in cancer treatment, immunotherapy has reached notable achievements, especially with the PD-L1 blockade. However, the efficacy of PD-L1 inhibitor therapy has been found to be limited due to activation of other immune checkpoints including TIM-3 and VISTA. So far, some studies have reported that the combination therapy targeting distinct types of immune checkpoints has been proved effective in several cancers.
5. Conclusions
As an inhibitor in the immune system, PD-L1 plays multiple roles in tumors. PD-L1 has been confirmed as a prospective and prognostic biomarker in certain cancers, while rebiopsy should be considered when PD-L1 expression is increased due to treatment (such as gefitinib treatment). Immune resistance induced by PD-L1 following various therapies inspired a combination therapy of PD-L1 blockade and these therapies. To date, immunotherapy, especially PD-1/PD-L1 blockade, which is at forefront of clinical therapy, has benefited many patients. However, primary and acquired resistance to this blockade therapy still exists and limits its efficacy. So far, key mechanisms suggest complement approaches for patients who cannot respond well to PD-1/PD-L1 antibodies. For example, modulating the immunosuppressive tumor microenvironment, such as depletion of Tregs, IDO, or MDSCs, interfering suppressive cytokines and inhibiting alternative immune checkpoints, may enhance the therapeutic efficacy of the PD-1/PD-L1 blockade. Other mechanisms underlying resistance to this blockade therapy and individual treatments for more patients requires further investigation.