Kidney transplantation is the preferred treatment for end-stage kidney disease (ESKD). Compared to maintenance dialysis, kidney transplantation results in improved patient survival and quality of life. Kidneys from living donors perform best; however, many patients with ESKD depend on kidneys from deceased donors. After procurement, donor kidneys are placed in a cold-storage solution until a suitable recipient is located. Sadly, prolonged cold storage times are associated with inferior transplant outcomes; therefore, in most situations when considering donor kidneys, long cold-storage times are avoided. The identification of novel mechanisms of cold-storage-related renal damage will lead to the development of new therapeutic strategies for preserving donor kidneys; to date, these mechanisms remain poorly understood. In this review, we discuss the importance of mitochondrial function, protein homeostasis, and renal recovery during stress from cold storage plus transplantation. Additionally, we discuss novel targets for therapeutic intervention.
Kidney transplantation is the preferred treatment for end-stage kidney disease (ESKD). Compared to maintenance dialysis, kidney transplantation results in improved patient survival and quality of life. Kidneys from living donors perform best; however, many patients with ESKD depend on kidneys from deceased donors. After procurement, donor kidneys are placed in a cold-storage solution until a suitable recipient is located. Sadly, prolonged cold storage times are associated with inferior transplant outcomes; therefore, in most situations when considering donor kidneys, long cold-storage times are avoided. The identification of novel mechanisms of cold-storage-related renal damage will lead to the development of new therapeutic strategies for preserving donor kidneys; to date, these mechanisms remain poorly understood. In this review, we discuss the importance of mitochondrial function, protein homeostasis, and renal recovery during stress from cold storage plus transplantation. Additionally, we discuss novel targets for therapeutic intervention.
- cold storage
- transplantation
- proteasome function
- mitochondrial function
- therapetutics
1. Introduction
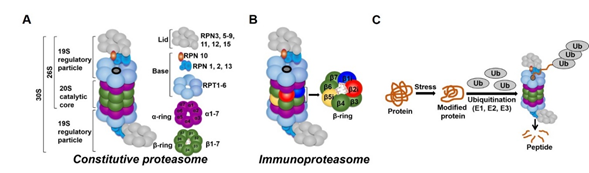
In all tissues, proteasomes are crucial for degrading modified, misfolded, and damaged proteins. The constitutive proteasome, selectively degrades ubiquitinated proteins (via the concerted actions of ubiquitinating enzymes) to small peptides (Figure 2A and [136–139]). The constitutive proteasome, a multi-subunit holoenzyme of ~2.5 MDa, is made up of two distinct sub-domains, namely, a 20S catalytic core particle and 1 or 2 19S regulatory particle(s) (Figure 2A). The 20S core particle is a barrel-shaped complex, composed of stacks of 2 β-rings (each ring made up of β1-7 subunits) in the center and 2 α-rings (each ring made up of α 1-7 subunits) on each end (Figure 2A). The α-rings (subunits) appear to have a regulatory function, allowing only unfolded substrates to enter into the 20S catalytic core. The β-ring of the 20S proteasome has 3 to 7 active sites (β-catalytic subunits) (Figure 2A) that hydrolyze peptide bonds in a chymotrypsin-like (β5 subunit), trypsin-like (β2 subunit), or caspase-like (β1 subunit) fashion (Figure 2C and [140]). The 19S regulatory particle recognizes and unfolds the ubiquitinated substrates before allowing the substrate to enter the 20S pore [141]. Functionally, the 19S particle is divided into a base and a lid. The base consists of an ATPase ring, made up of 6 AAA-ATPase subunits (Rpt1-6), and 3 non-ATPase subunits (Rpn1, 2, and 13) (Figure 2A). The ATPase subunits consume ATP to unfold the substrate and help translocate it to the pore of the 20S catalytic core. The lid, which is linked to the base by the Rpn10 subunit (Figure 2A), assists in the efficient degradation of the ubiquitinated substrates. The immunoproteasome is a proteasome variant that is normally found in immune cell compartments [142]. However, in response to inflammation, catalytic subunits (β1, β2, and β5) of the constitutive proteasome are exchanged for immunoproteasome subunits (β1i, β2i, and β5i) in most non-immune cells (Figure 2B and [142,143]).
Acute kidney injury affects approximately 13–18% of hospitalized patients and has been shown to be associated with increased mortality. [1]. Additionally, acute kidney injury has also been linked to the development of chronic kidney disease and end-stage kidney disease (ESKD). ESKD affects 630,000 Americans and is the ninth-leading cause of death in the US (NIDDK, 2014). Kidney transplantation is the preferred treatment to increase longevity and quality of life for people with ESKD, but due to a shortage of transplantable kidneys, 7 of 10 ESKD patients will remain on dialysis (and many will die) while waiting for a kidney (95,268 waiting-list candidates vs. 19,848 transplants in 2017; http://www.unos.org).
Advances in tissue-type matching and immunosuppressive protocols have greatly reduced the incidence of acute transplant rejection and short-term graft dysfunction; however, optimizing long-term graft function continues to be a challenge, especially with kidneys from deceased donors. Kidneys from living donors have better long-term graft outcomes than those from deceased donors. One of the key variables is cold storage (CS) [2][3][4], which is the universal method for preserving kidneys from donors that allows time for identification of potential recipients, transportation of kidneys, tissue typing, and cross-matching. Kidneys from living donors generally are exposed to only a few hours of CS, while those from deceased donors undergo long hours of CS during transport, tissue typing, and cross-matching. Despite the increasing use of hypothermic machine perfusion (HMP) for the preservation of deceased donor kidneys, most transplant centers still rely on static CS techniques. Both CS techniques (HMP or static) lower the metabolic rate, allowing the organ to be stored until a recipient is located [5][6]. Acceptable CS times vary between transplant centers, ranging from 24 to 72 h. Unfortunately, each additional hour of CS increases the risk of graft failure [2][3][4] via poorly described mechanisms. Tragically, approximately 20% of donor kidneys that are retrieved each year are discarded or not transplanted (http://www.unos.org), partly due to prolonged CS [7][8]. A better understanding of injury-related pathways secondary to CS, and the identification of novel therapies aimed to mitigate damage would likely lead to improved deceased donor transplant rates and patient outcomes.
The kidney is a composite organ made up of many blood-filtering units called nephrons. Each nephron consists of anatomically and functionally discrete segments known as a renal corpuscle, proximal tubule, loop of Henle, distal tubule, and collecting duct system [9]. Each segment of the nephron is composed of multiple cell types of both epithelial and mesenchymal origin; all of which participate in various functions such as removing nitrogen waste and other waste products, controlling blood electrolytes and acid-base balance, and secreting hormones that regulate blood composition and blood pressure [9][10][11][12][13]. Like most organs, renal tissues consume a high level of oxygen [14][15][16], and have quality-control mechanisms that help fold nascent polypeptides, clear unfolded or misfolded proteins, respond to protein aggregates, and dispose of potentially toxic molecules. The balance of energy and the integrity of the proteome is extremely important for renal cell viability during stressful conditions, especially during renal cold storage plus transplantation (CS/Tx). One could reasonably expect to see damage within various compartments of renal tissue when the kidney undergoes a series of stressors, namely CS and blood reperfusion events during transplantation. Recovery from CS/Tx-induced stress is possible when the repair process remains in balance and overcomes the rate of CS/Tx-mediated injury. Unfortunately, very little is known about the mechanisms of CS/Tx-mediated damage that leads to renal dysfunction after CS/Tx. Here, we will discuss some of the literature that describes the triggers of damage during renal transplantation. Specifically, we will discuss the importance of mitochondrial and proteasomal function to maintain mitochondrial protein quality during renal CS/Tx and potential strategies to prevent organ damage and improve transplant outcomes.
2. The Proteasome
In all tissues, proteasomes are crucial for degrading modified, misfolded, and damaged proteins. The constitutive proteasome, selectively degrades ubiquitinated proteins (via the concerted actions of ubiquitinating enzymes) to small peptides (Figure 1A and [17][18][19][20]). The constitutive proteasome, a multi-subunit holoenzyme of ~2.5 MDa, is made up of two distinct sub-domains, namely, a 20S catalytic core particle and 1 or 2 19S regulatory particle(s) (Figure 1A). The 20S core particle is a barrel-shaped complex, composed of stacks of 2 β-rings (each ring made up of β1-7 subunits) in the center and 2 α-rings (each ring made up of α 1-7 subunits) on each end (Figure 1A). The α-rings (subunits) appear to have a regulatory function, allowing only unfolded substrates to enter into the 20S catalytic core. The β-ring of the 20S proteasome has 3 to 7 active sites (β-catalytic subunits) (Figure 1A) that hydrolyze peptide bonds in a chymotrypsin-like (β5 subunit), trypsin-like (β2 subunit), or caspase-like (β1 subunit) fashion (Figure 1C and [21]). The 19S regulatory particle recognizes and unfolds the ubiquitinated substrates before allowing the substrate to enter the 20S pore [22]. Functionally, the 19S particle is divided into a base and a lid. The base consists of an ATPase ring, made up of 6 AAA-ATPase subunits (Rpt1-6), and 3 non-ATPase subunits (Rpn1, 2, and 13) (Figure 1A). The ATPase subunits consume ATP to unfold the substrate and help translocate it to the pore of the 20S catalytic core. The lid, which is linked to the base by the Rpn10 subunit (Figure 1A), assists in the efficient degradation of the ubiquitinated substrates. The immunoproteasome is a proteasome variant that is normally found in immune cell compartments [23]. However, in response to inflammation, catalytic subunits (β1, β2, and β5) of the constitutive proteasome are exchanged for immunoproteasome subunits (β1i, β2i, and β5i) in most non-immune cells (Figure 1B and [23][24]).
Figure 12.
Ubiquitin-proteasome system (UPS). (
A
) The constitutive proteasome (26S or 36S) is a barrel-shaped organelle that is made up of 20S catalytic core and one (26S) or two (30S) 19S particle(s). The 20S catalytic core is made up of stacks of two β rings (β1–β7) and two α rings (α1–α7). The 19S regulatory particle is made up of a lid and a base with multiple subunits. (
B
) The immunoproteasome is a proteasome variant that is normally found in immune cell compartments. However, in response to inflammation, constitutive proteasome subunits (β1, β2, and β5) are exchanged for the immunoproteasome subunits (β1i, β2i, and β5i) in most non-immune cells in the body. (
C
) Damaged or modified proteins are ubiquitinated (with the concerted action of ubiquitinating enzymes), which is then recognized by the constitutive proteasome for degradation. The constitutive proteasome selectively degrades ubiquitinated proteins to small peptides (
A
); it has 3 to 7 protease active sites (β-catalytic subunits) that hydrolyze peptide bonds in a chymotrypsin (β5 subunit)-, trypsin (β2 subunit)-, or caspase (β1 subunit)-like fashion. Following protein degradation, the peptides are released and recycled.
The proteasome maintains functional protein homeostasis, also known as proteostasis, by monitoring misfolded and damaged proteins; however, this is a challenge in the context of renal CS/Tx, especially in kidneys that have undergone prolonged CS (Figure 2). Given that prolonged CS followed by warm IRI produces ROS [25][26], and that ROS modulate the constitutive proteasome function [27][28][29], we can postulate that CS/Tx-mediated ROS could trigger denaturation of intracellular proteins and modulation of the constitutive proteasome function. Indeed, a recent report demonstrated that the chymotrypsin-like activity of the proteasome was compromised after renal CS/Tx [30], and that this correlated with severe renal dysfunction [31][32]. A study performed by using pharmacological inhibition of chymotrypsin-like activity of the proteasome during warm IRI showed aggravated renal damage [33]. Genetic or pharmacologic modulation of proteasome function (chymotrypsin-like) inhibition, achieved by siRNA or bortezomib treatment, respectively, in rat proximal tubular cells showed increase of ROS production [34]. At this point, the mechanisms of proteasome dysfunction during CS/Tx are not known. One of the possible mechanisms could be CS/Tx-mediated post-translational modification of the proteasome subunits because this mechanism has been described to modulate proteasome function and assembly in various experimental models [35][36].
The proteasome maintains functional protein homeostasis, also known as proteostasis, by monitoring misfolded and damaged proteins; however, this is a challenge in the context of renal CS/Tx, especially in kidneys that have undergone prolonged CS (Figure 3). Given that prolonged CS followed by warm IRI produces ROS [37,39], and that ROS modulate the constitutive proteasome function [144–146], we can postulate that CS/Tx-mediated ROS could trigger denaturation of intracellular proteins and modulation of the constitutive proteasome function. Indeed, a recent report demonstrated that the chymotrypsin-like activity of the proteasome was compromised after renal CS/Tx [147], and that this correlated with severe renal dysfunction [80,90]. A study performed by using pharmacological inhibition of chymotrypsin-like activity of the proteasome during warm IRI showed aggravated renal damage [148]. Genetic or pharmacologic modulation of proteasome function (chymotrypsin-like) inhibition, achieved by siRNA or bortezomib treatment, respectively, in rat proximal tubular cells showed increase of ROS production [149]. At this point, the mechanisms of proteasome dysfunction during CS/Tx are not known. One of the possible mechanisms could be CS/Tx-mediated post-translational modification of the proteasome subunits because this mechanism has been described to modulate proteasome function and assembly in various experimental models [150,151].
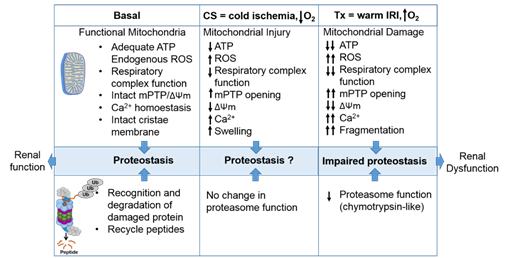
Figure 23. The intricate relationship between the mitochondria and proteasome. Schematic summary depicting mitochondrial and proteasomal changes during cold storage (CS) and transplantation. During CS, mitochondrial respiration function, ATP level, and mitochondrial membrane potential (ΔΨm) decreases, whereas ROS and calcium levels increase leading to an increase of mitochondrial permeability transition pore (mPTP) opening. This leads to increased swelling and decreased function of mitochondria. These changes are further exacerbated and sustained following transplantation leading to mitochondrial fragmentation and bioenergetic crisis. Proteasome function remains unchanged during renal CS, whereas the chymotrypsin-like proteasome function is decreased following transplantation. This leads to alteration of mitochondrial protein homeostasis and acute tubular necrosis after transplantation and significantly decreases renal function.
The intricate relationship between the mitochondria and proteasome. Schematic summary depicting mitochondrial and proteasomal changes during cold storage (CS) and transplantation. During CS, mitochondrial respiration function, ATP level, and mitochondrial membrane potential (ΔΨm) decreases, whereas ROS and calcium levels increase leading to an increase of mitochondrial permeability transition pore (mPTP) opening. This leads to increased swelling and decreased function of mitochondria. These changes are further exacerbated and sustained following transplantation leading to mitochondrial fragmentation and bioenergetic crisis. Proteasome function remains unchanged during renal CS, whereas the chymotrypsin-like proteasome function is decreased following transplantation. This leads to alteration of mitochondrial protein homeostasis and acute tubular necrosis after transplantation and significantly decreases renal function.
It is well-accepted that CS/Tx produces inflammation and releases inflammatory cytokines [152]. Immunoproteasomes are induced and activated in response to the inflammatory cytokines, and help in processing donor-derived antigen effectively. Unlike the constitutive proteasome, the immunoproteasome is resistant to oxidative stress and can function in an ATP-independent manner [153,154]. For example, interferon-induced ROS activate the immunoproteasome [154]. There are a handful of transplant studies showing a negative correlation of immunoproteasome activity with organ function. In this context, a recent report by Li et al. indicates that pharmacological inhibition of the immunoproteasome with ONX 0914 (a reversible β5i inhibitor) reduces donor-specific antibody production in a rat model of renal CS/Tx [155]. Thus, it is worth investigating whether ONX 0914 should be included in the CS solution. This is particularly exciting because some studies indicate that ONX 0914 protects against cardiac and neuronal IRI [156,157]. However, one caveat of the Li et al. study is that the authors used a very short CS time (~35 min) that is likely not clinically relevant. Future studies with longer CS times are needed to verify whether blunting the immunoproteasome with ONX 0914 during CS confers protection after transplantation. The specific mechanisms underlying compromised proteasome function and exacerbated immunoproteasome activity during renal CS/Tx are not understood and should be addressed.
Although the proteasome manages protein turnover, aberrations in the expression and function of the constitutive proteasome and immunoproteasome are implicated in the pathogenesis of several human diseases, including cancer, autoimmune disorders, and inflammatory diseases [142,143,158–161]. In the context of CS/Tx, it is clear that the initial damage occurs within the epithelial or vascular compartments within the kidneys (during cold ischemia) that eventually triggers an immune response following blood reperfusion (warm IRI) after CS/Tx. It is expected that prolonged CS followed by transplantation disrupts protein homeostasis, which overwhelms the immune response, and this could directly impact long-term graft and patient outcomes.
It is well-accepted that CS/Tx produces inflammation and releases inflammatory cytokines [37]. Immunoproteasomes are induced and activated in response to the inflammatory cytokines, and help in processing donor-derived antigen effectively. Unlike the constitutive proteasome, the immunoproteasome is resistant to oxidative stress and can function in an ATP-independent manner [38][39]. For example, interferon-induced ROS activate the immunoproteasome [39]. There are a handful of transplant studies showing a negative correlation of immunoproteasome activity with organ function. In this context, a recent report by Li et al. indicates that pharmacological inhibition of the immunoproteasome with ONX 0914 (a reversible β5i inhibitor) reduces donor-specific antibody production in a rat model of renal CS/Tx [40]. Thus, it is worth investigating whether ONX 0914 should be included in the CS solution. This is particularly exciting because some studies indicate that ONX 0914 protects against cardiac and neuronal IRI [41][42]. However, one caveat of the Li et al. study is that the authors used a very short CS time (~35 min) that is likely not clinically relevant. Future studies with longer CS times are needed to verify whether blunting the immunoproteasome with ONX 0914 during CS confers protection after transplantation. The specific mechanisms underlying compromised proteasome function and exacerbated immunoproteasome activity during renal CS/Tx are not understood and should be addressed.
Although the proteasome manages protein turnover, aberrations in the expression and function of the constitutive proteasome and immunoproteasome are implicated in the pathogenesis of several human diseases, including cancer, autoimmune disorders, and inflammatory diseases [23][24][43][44][45][46]. In the context of CS/Tx, it is clear that the initial damage occurs within the epithelial or vascular compartments within the kidneys (during cold ischemia) that eventually triggers an immune response following blood reperfusion (warm IRI) after CS/Tx. It is expected that prolonged CS followed by transplantation disrupts protein homeostasis, which overwhelms the immune response, and this could directly impact long-term graft and patient outcomes.
- Shrum, S.; MacMillan-Crow, L.A.; Parajuli, N. Cold Storage Exacerbates Renal and Mitochondrial Dysfunction Following Transplantation. J. Kidney 2016, 2. [Google Scholar] [CrossRef]
- Pajuri, N.; MacMillan-Crow, L.A. Role of reduced manganese superoxide dismutase in ischemia-reperfusion injury: A possible trigger for autophagy and mitochondrial biogenesis? Am. J. Physiol. Physiol. 2012, 304, 257–267. [Google Scholar] [CrossRef]
- Parajuli, N.; Marine, A.; Simmons, S.; Saba, H.; Mitchell, T.; Shimizu, T.; Shirasawa, T.; MacMillan-Crow, L.A. Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse. Free Radic. Boil. Med. 2011, 51, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Kosieradzki, M.; Rowiński, W. Ischemia/Reperfusion Injury in Kidney Transplantation: Mechanisms and Prevention. Transplant. Proc. 2008, 40, 3279–3288. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Cellular and molecular derangements in acute tubular necrosis. Curr. Opin. Pediatr. 2005, 17, 193–199. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Boil. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Wang, X.; Ding, R.; Tao, T.-Q.; Mao, H.-M.; Liu, M.; Xie, Y.-S.; Liu, X. Myofibrillogenesis Regulator 1 Rescues Renal Ischemia/Reperfusion Injury by Recruitment of PI3K-Dependent P-AKT to Mitochondria. Shock 2016, 46, 531–540. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Nakagawa, T.; Shimizu, S. Mitochondrial membrane permeability transition and cell death. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1297–1300. [Google Scholar] [CrossRef]
- Atkinson, S.J.; Hosford, M.A.; Molitoris, B.A. Mechanism of Action Polymerization in Cellular ATP Depletion. J. Boil. Chem. 2003, 279, 5194–5199. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Morrison, A.R.; Pascoe, N.; Tauk, N.; Kennerly, D. Biochemical alterations of membrane lipids associated with renal injury. Fed. Proc. 1984, 43, 2811–2814. [Google Scholar]
- Arnarez, C.; Mazat, J.-P.; Elezgaray, J.; Marrink, S.J.; Periole, X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Ball, W.B.; Neff, J.K.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2017, 592, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Boil. Chem. 2005, 280, 29403–29408. [Google Scholar] [CrossRef] [PubMed]
- Mehdipour, A.R.; Hummer, G. Cardiolipin puts the seal on ATP synthase. Proc. Nat. Acad. Sci. USA 2016, 113, 8568–8570. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.; Robinson, A.J.; Walker, J.E. Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases. Proc. Nat. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef] [PubMed]
- Vasdev, S.C.; Biro, G.P.; Narbaitz, R.; Kako, K.J. Membrane changes induced by early myocardial ischemia in the dog. Can. J. Biochem. 1980, 58, 1112–1119. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Minkler, P.; Hoppel, C.L. Enhanced modification of cardiolipin during ischemia in the aged heart. J. Mol. Cell. Cardiol. 2009, 46, 1008–1015. [Google Scholar] [CrossRef]
- Wiswedel, I.; Gardemann, A.; Storch, A.; Peter, D.; Schild, L. Degradation of phospholipids by oxidative stress--exceptional significance of cardiolipin. Free Radic. Res. 2010, 44, 135–145. [Google Scholar] [CrossRef]
- Petrosillo, G.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Paradies, G. Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: Induction of permeability transition and cytochromecrelease. FEBS Lett. 2006, 580, 6311–6316. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Ca2+-induced Reactive Oxygen Species Production Promotes CytochromecRelease from Rat Liver Mitochondria via Mitochondrial Permeability Transition (MPT)-dependent and MPT-independent Mechanisms. J. Boil. Chem. 2004, 279, 53103–53108. [Google Scholar] [CrossRef]
- Silvestro, L.; Ruikun, C.; Sommer, F.; Duc, T.; Biancone, L.; Montrucchio, G.; Camussi, G. Platelet-Activating Factor-Induced Endothelial Cell Expression of Adhesion Molecules and Modulation of Surface Glycocalyx, Evaluated by Electron Spectroscopy for Chemical Analysis. Semin. Thromb. Hemost. 1994, 20, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, A.; Matsukuma, Y.; Ueki, K.; Nishiki, T.; Doi, A.; Okabe, Y.; Nakamura, M.; Tsuruya, K.; Nakano, T.; Kitazono, T.; et al. Thrombotic microangiopathy associated with anticardiolipin antibody in a kidney transplant recipient with polycythemia. CEN Case Rep. 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-X.; Yin, X.-M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Boil. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, H.; Zhang, Y.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mammalian STE20-Like Kinase 1 Deletion Alleviates Renal Ischaemia-Reperfusion Injury via Modulating Mitophagy and the AMPK-YAP Signalling Pathway. Cell. Physiol. Biochem. 2018, 51, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Han, F.; Xiang, H.; Wang, Y.; Dou, M.; Xia, X.; Li, Y.; Zheng, J.; Ding, X.; Xue, W.; et al. Role of prostaglandin E2 receptor 4 in the modulation of apoptosis and mitophagy during ischemia/reperfusion injury in the kidney. Mol. Med. Rep. 2019, 20, 3337–3346. [Google Scholar] [CrossRef]
- Ang, C.T.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia–reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Jackson, E.K.; Menshikova, E.V.; Mi, Z.; Verrier, J.D.; Bansal, R.; Janesko-Feldman, K.; Jackson, T.C.; Kochanek, P.M. Renal 2’,3’-Cyclic Nucleotide 3’-Phosphodiesterase Is an Important Determinant of AKI Severity after Ischemia-Reperfusion. J. Am. Soc. Nephrol. JASN 2016, 27, 2069–2081. [Google Scholar] [CrossRef]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef] [PubMed]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.-M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [PubMed]
- De Deken, J.; Rex, S.; Lerut, E.; Martinet, W.; Monbaliu, D.; Pirenne, J.; Jochmans, I. Postconditioning effects of argon or xenon on early graft function in a porcine model of kidney autotransplantation. BJS 2018, 105, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Keys, D.; Nydam, T.; Plenter, R.J.; Edelstein, C.L.; Jani, A. Inhibition of autophagy increases apoptosis during re-warming after cold storage in renal tubular epithelial cells. Transpl. Int. 2014, 28, 214–223. [Google Scholar] [CrossRef]
- Pallet, N. Emerging Roles of Autophagy in the Stressed Kidney Allograft. Semin. Nephrol. 2014, 34, 34–41. [Google Scholar] [CrossRef]
- Pallet, N.; Livingston, M.; Dong, Z. Emerging Functions of Autophagy in Kidney Transplantation. Arab. Archaeol. Epigr. 2013, 14, 13–20. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Damage-associated molecular patterns derived from mitochondria may contribute to the hemodialysis-associated inflammation. Int. Urol. Nephrol. 2013, 46, 107–112. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Wu, J.; Li, G.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; et al. Urinary Mitochondrial DNA Levels Identify Acute Kidney Injury in Surgical Critical Illness Patients. Shock 2017, 48, 11–17. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Ren, H.; Wu, J.; Wu, X.; Liu, S.; Wang, G.; Gu, G.; Guo, K.; Li, J. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxidative Med. Cell. Longev. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Kim, K.; Moon, H.; Lee, Y.H.; Seo, J.-W.; Kim, Y.G.; Moon, J.-Y.; Kim, J.S.; Jeong, K.-H.; Lee, T.W.; Ihm, C.-G.; et al. Clinical relevance of cell-free mitochondrial DNA during the early postoperative period in kidney transplant recipients. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Soares, F.; Tattoli, I.; Girardin, S.E. Mitochondria in innate immunity. EMBO Rep. 2011, 12, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2010, 12, 222–230. [Google Scholar] [CrossRef]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2015, 68, 20–48. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-dependent proteolytic pathway: Specificity of recognition of the proteolytic substrates. Revis. Sobre Boil. Cel. RBC 1989, 20, 217–234. [Google Scholar]
- Glickman, M.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Zwickl, P.; Voges, D.; Baumeister, W. The proteasome: A macromolecular assembly designed for controlled proteolysis. Philos. Trans. R. Soc. B: Boil. Sci. 1999, 354, 1501–1511. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The Ubiquitin Pathway for the Degradation of Intracellular Proteins. Prog. Nucl. Acid Res. Mol. Biol. 1986, 33, 19–56. [Google Scholar] [CrossRef]
- Jung, T.; Grune, T. Structure of the Proteasome. Prog. Mol. Biol. Transl. Sci. 2012, 109, 1–39. [Google Scholar] [CrossRef]
- Lander, G.C.; Estrin, E.; Matyskiela, M.E.; Bashore, C.; Nogales, E.; Martin, A. Complete subunit architecture of the proteasome regulatory particle. Nature 2012, 482, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33. [Google Scholar]
- Kaur, G.; Batra, S. Emerging role of immunoproteasomes in pathophysiology. Immunol. Cell Boil. 2016, 94, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Sjöstrand, J.; Karlsson, J.-O. Differential Inhibition of Three Peptidase Activities of the Proteasome in Human Lens Epithelium by Heat and Oxidation. Exp. Eye Res. 1999, 69, 129–138. [Google Scholar] [CrossRef]
- Reinheckel, T.; Sitte, N.; Ullrich, O.; Kuckelkorn, U.; Davies, K.E.; Grune, T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem. J. 1998, 335, 637–642. [Google Scholar] [CrossRef]
- Reinheckel, T.; Ullrich, O.; Sitte, N.; Grune, T. Differential Impairment of 20S and 26S Proteasome Activities in Human Hematopoietic K562 Cells during Oxidative Stress. Arch. Biochem. Biophys. 2000, 377, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.; MacMillan-Crow, L.A.; Parajuli, N. Renal cold storage followed by transplantation impairs proteasome function and mitochondrial protein homeostasis. Am. J. Physiol. 2019, 316, 42–53. [Google Scholar] [CrossRef]
- Huber, J.M.; Tagwerker, A.; Heininger, D.; Mayer, G.; Rosenkranz, A.R.; Eller, K. The proteasome inhibitor Bortezomib aggravates renal ischemia-reperfusion injury. Am. J. Physiol. 2009, 297, 451–460. [Google Scholar] [CrossRef]
- Parajuli, N. A Cycle of Altered Proteasome and Reactive Oxygen Species Production in Renal Proximal Tubular Cells. Toxicol. Forensic Med. Open J. 2019, 4, 13–17. [Google Scholar] [CrossRef]
- Scruggs, S.B.; Zong, N.C.; Wang, D.; Stefani, E.; Ping, P. Post-translational modification of cardiac proteasomes: Functional delineation enabled by proteomics. Am. J. Physiol. Circ. Physiol. 2012, 303, 9–18. [Google Scholar] [CrossRef]
- Kors, S.; Geijtenbeek, K.; Reits, E.; Schipper-Krom, S. Regulation of Proteasome Activity by (Post-)transcriptional Mechanisms. Front. Mol. Biosci. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, A.; Mehta, R.; Sood, P.; Hariharan, S. Early allograft inflammation and scarring associate with graft dysfunction and poor outcomes in renal transplant recipients with delayed graft function: A prospective single center cohort study. Transpl. Int. 2018, 31, 1369–1379. [Google Scholar] [CrossRef]
- Aiken, C.T.; Kaake, R.M.; Wang, X.; Huang, L. Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteom. MCP 2011, 10. [Google Scholar]
- Seifert, U.; Bialy, L.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Basler, M.; Alvarez, G.; Brunner, T.; Kirk, C.J.; Groettrup, M. Immunoproteasome inhibition prevents chronic antibody-mediated allograft rejection in renal transplantation. Kidney Int. 2018, 93, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.; Wang, Y.; Lei, H.; Su, H.; Zeng, J.; Pei, Z.; Huang, R. Inhibition of immunoproteasome reduces infarction volume and attenuates inflammatory reaction in a rat model of ischemic stroke. Cell Death Dis. 2015, 6, 1626. [Google Scholar] [CrossRef]
- Karreci, E.S.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc. Nat. Acad. Sci. USA 2016, 113, 8425–8432. [Google Scholar] [CrossRef]
- Bellavista, E.; Andreoli, F.; Parenti, M.D.; Martucci, M.; Santoro, A.; Salvioli, S.; Capri, M.; Baruzzi, A.; Del Rio, A.; Franceschi, C.; et al. Immunoproteasome in Cancer and Neuropathologies: A New Therapeutic Target? Curr. Pharm. Des. 2013, 19, 702–718. [Google Scholar] [CrossRef]
- Miller, Z.; Ao, L.; Kim, K.B.; Lee, W. Inhibitors of the immunoproteasome: Current status and future directions. Curr. Pharm. Des. 2013, 19, 4140–4151. [Google Scholar] [CrossRef]
- Paul, S. Dysfunction of the ubiquitin-proteasome system in multiple disease conditions: Therapeutic approaches. BioEssays 2008, 30, 1172–1184. [Google Scholar] [CrossRef]
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. et Biophys. Acta Bioenerg. 2013, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Glickman, M. Ubiquitin–Proteasome System and mitochondria—Reciprocity. Biochim. Biophys. Acta Bioenerg. 2011, 1809, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The Ubiquitin-Proteasome System Regulates Mitochondrial Intermembrane Space Proteins. Mol. Cell. Boil. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Chander, H.; Schäfer, P.; Meiss, G.; Krüger, R.; Schulz, J.B.; Germain, D. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Boil. Chem. 2008, 283, 12681–12685. [Google Scholar] [CrossRef]
- Jeon, H.B.; Choi, E.S.; Yoon, J.H.; Hwang, J.H.; Chang, J.W.; Lee, E.K.; Choi, H.W.; Park, Z.-Y.; Yoo, Y.J. A proteomics approach to identify the ubiquitinated proteins in mouse heart. Biochem. Biophys. Res. Commun. 2007, 357, 731–736. [Google Scholar] [CrossRef]
- Altmann, K.; Westermann, B. Role of Essential Genes in Mitochondrial Morphogenesis In Saccharomyces cerevisiae. Mol. Boil. Cell 2005, 16, 5410–5417. [Google Scholar] [CrossRef]
- Azzu, V.; Brand, M.D. Degradation of an intramitochondrial protein by the cytosolic proteasome. J. Cell Sci. 2010, 123, 578–585. [Google Scholar] [CrossRef]
- Neutzner, A.; Youle, R.J.; Karbowski, M. Outer mitochondrial membrane protein degradation by the proteasome. Ciba Found. Sym. Bilharz. 2007, 287, 4–20. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr. Opin. Cell Boil. 2011, 23, 476–482. [Google Scholar] [CrossRef]
- Chatenay-Lapointe, M.; Shadel, G.S. Stressed-Out Mitochondria Get MAD. Cell Metab. 2010, 12, 559–560. [Google Scholar] [CrossRef]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Boil. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Brand, M.D. Characteristics of the turnover of uncoupling protein 3 by the ubiquitin proteasome system in isolated mitochondria. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Mookerjee, S.A.; Brand, M.D. Rapid turnover of mitochondrial uncoupling protein 3. Biochem. J. 2010, 426, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Geng, Q.; Romero, J.; Saini, V.; Baker, T.A.; Picken, M.M.; Gamelli, R.L.; Majetschak, M. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem. Biophys. Res. Commun. 2009, 390, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, X.; Li, S.; Liu, N.; Lian, W.; McDowell, E.; Zhou, P.; Zhao, C.; Guo, H.; Zhang, C.; et al. Physiological levels of ATP negatively regulate proteasome function. Cell Res. 2010, 20, 1372–1385. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R.; Davies, K.E.; Divald, A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J. Mol. Cell. Cardiol. 2006, 42, 265–269. [Google Scholar] [CrossRef]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.; Finley, D. A gated channel into the proteasome core particle. Nat. Genet. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Bech-Otschir, D.; Helfrich, A.; Enenkel, C.; Consiglieri, G.; Seeger, M.; Holzhütter, H.-G.; Dahlmann, B.; Kloetzel, P.-M. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat. Struct. Mol. Boil. 2009, 16, 219–225. [Google Scholar] [CrossRef]
- Smith, D.; Benaroudj, N.; Goldberg, A.L. Proteasomes and their associated ATPases: A destructive combination. J. Struct. Boil. 2006, 156, 72–83. [Google Scholar] [CrossRef]
- Smith, D.; Fraga, H.; Reis, C.; Kafri, G.; Goldberg, A.L. ATP Binds to Proteasomal ATPases in Pairs with Distinct Functional Effects, Implying an Ordered Reaction Cycle. Cell 2011, 144, 526–538. [Google Scholar] [CrossRef]
- Kron, P.; Schlegel, A.; de Rougemont, O.; Oberkofler, C.E.; Clavien, P.A.; Dutkowski, P. Short Cool, and Well Oxygenated-HOPE for Kidney Transplantation in a Rodent Model. Ann. Surg. 2016, 264, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Kron, P.; Schlegel, A.; Muller, X.; Gaspert, A.; Clavien, P.-A.; Dutkowski, P. Hypothermic Oxygenated Perfusion. Transplantation 2019, 103, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yen, J.; Kaiser, P.; Huang, L. Regulation of the 26S Proteasome Complex During Oxidative Stress. Sci. Signal. 2010, 3, 88. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Grune, T. The proteasome and the degradation of oxidized proteins: Part I-structure of proteasomes. Redox Boil. 2013, 1, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II -protein oxidation and proteasomal degradation. Redox Boil. 2013, 2, 99–104. [Google Scholar] [CrossRef]
- Sitte, N.; Merker, K.; Grune, T. Proteasome-dependent degradation of oxidized proteins in MRC-5 fibroblasts. FEBS Lett. 1998, 440, 399–402. [Google Scholar] [CrossRef]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.; Bradley, J.A.; Pettigrew, G.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted antioxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Hear. Lung Transplant. 2015, 34, 1471–1480. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.; Pettigrew, G.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Boil. 2015, 5, 163–168. [Google Scholar] [CrossRef]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta Bioenerg. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.K.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Talbert, E.E.; Smuder, A.J.; Hudson, M.; Nelson, W.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Powers, S.K. A Mitochondrial-targeted Antioxidant Protects against Mechanical Ventilation-induced Diaphragm Weakness. Med. Sci. Sports Exerc. 2010, 42, 17–18. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Liu, S.; Soong, Y.; Szeto, H.H. Disruption of cytochrome c heme coordination is responsible for mitochondrial injury during ischemia. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1075–1084. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochromec/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Saad, A.; Herrmann, S.M.; Eirin, A.; Ferguson, C.M.; Glockner, J.F.; Bjarnason, H.; McKusick, M.A.; Misra, S.; Lerman, L.O.; Textor, S.C. Phase 2a Clinical Trial of Mitochondrial Protection (Elamipretide) During Stent Revascularization in Patients With Atherosclerotic Renal Artery Stenosis. Circ. Cardiovasc. Interv. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology 2003, 124, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates someznecrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Kim, S.Y.; Shim, M.S.; Kim, K.-Y.; Weinreb, R.N.; A Wheeler, L.; Ju, W.-K. Inhibition of cyclophilin D by cyclosporin A promotes retinal ganglion cell survival by preventing mitochondrial alteration in ischemic injury. Cell Death Dis. 2014, 5, 1105. [Google Scholar] [CrossRef]
- Singh, D.; Chander, V.; Chopra, K. Cyclosporine protects against ischemia/reperfusion injury in rat kidneys. Toxicology 2005, 207, 339–347. [Google Scholar] [CrossRef]
- De Arriba, G.; Calvino, M.; Benito, S.; Parra, T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol. Lett. 2013, 218, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Puigmulé, M.; Lopez-Hellin, J.; Suñè, G.; Tornavaca, O.; Camano, S.; Tejedor, A.; Meseguer, A. Differential proteomic analysis of cyclosporine A-induced toxicity in renal proximal tubule cells. Nephrol. Dial. Transplant. 2009, 24, 2672–2686. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.; Kukla, A.; Ibrahim, H.N. Calcineurin Inhibitor Nephrotoxicity: A Review and Perspective of the Evidence. Am. J. Nephrol. 2013, 37, 602–612. [Google Scholar] [CrossRef] [PubMed]