The mechanisms of acute respiratory failure other than inflammation and complicating the SARS-CoV-2 infection are still far from being fully understood, thus challenging the management of COVID-19 patients in the critical care setting. In this unforeseen scenario, the role of an indi-vidual’s excessive spontaneous breathing may acquire critical importance, being one potential and important driver of lung injury and disease progression.
- SARS-CoV-2
- COVID-19
- mechanical ventilation
- spontaneous breathing
- acute respiratory dis-tress syndrome
- acute respiratory failure.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
In the early phase of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) outbreak, the disproportionate number of patients with severe SARS-CoV-2 disease (COVID-19) and associated acute hypoxic respiratory failure (ARF), compared to the available resources forced clinicians to assist patients with ARF by non-invasive techniques outside intensive care units (ICU), keeping spontaneous breathing preserved despite dramatically impaired gas exchanges [1][2][1,2]. Parallelly, when invasive mechanical ventilation (MV) was prompted after non-invasive ventilation failure, a lack of substantial improvement was reported in a significant number of cases [2]. In this scenario, different phenotypes of lung damage have been speculated, starting from the host-driven exaggerated inflammatory response (“cytokine storm”) that may contribute to acute lung injury [2][3][2,3]. Furthermore, the intravascular coagulation activation seems to trigger the most severe evolution of COVID-19 and to interfere with the mechanisms of lung repair and wound healing, thus predisposing individuals to aberrant mechanisms of repair and fibrosis [4][5][6][4–6]. However, the pathophysiology of COVID-19-induced lung damages may not be limited to the inflammatory and micro-thrombotic hypotheses. Indeed, a recent exploratory study on 39 patients with COVID-19-related ARDS suggested that the hyperinflammatory phenotype is less prevalent, although more severe, in COVID-19 patients than in previous non-COVID-19 cohorts [7]. Thus, in patients with preserved spontaneous breathing, mechanical reasons beyond biochemical causes may be hypothesized in driving lung injury progression. In particular, the role of inspiratory effort in promoting lung damage phenotypes in COVID-19 is may be critical. With this perspective article, we tried to explore the dynamic interaction between spontaneous breathing and lung damage in the COVID-19 model of respiratory failure, forecasting the potential evolution of SARS-CoV-2 induced ARDS, in a new bio-mechanical phenotype of injured lungs.
2. COVID-19 and Phenotypes of Lung Damage
Based on physio-pathological hypotheses, patients with COVID-19 pneumonia undergoing mechanical ventilation can be divided into two major phenotypes: “Non-ARDS” type L (low elastance, low ventilation-to-perfusion ratio, low lung weight, low lung recruitability), and “ARDS” type H (high elastance, high right-to-left shunt, high lung weight, high lung recruitability) [1,3]. Type L seems to be the most frequent pattern and exhibits a dissociation between the mechanical characteristics of the respiratory system and the severity of hypoxemia. These patients show a radiological pattern characterized by ground-glass density with subpleural predominance, with only a slight increase in lung weight, normal lung compliance [8], and a loss of hypoxic vasoconstriction resulting in a low ventilation-to-perfusion (VA/Q) ratio [9]. Type H seems to exhibit typical ARDS features, with radiological appearance of bilateral consolidations, decreased compliance of the respiratory system, and increased lung weight [8][9][8,9]. Despite these observations, a recent study provides evidence that mechanically ventilated patients with COVID-19 display a form of lung injury similar to classical ARDS, and only 5–7% show static compliance greater than the 95th percentile of those with classical ARDS [9]. In line with these assumptions, the proposed COVID-19-related phenotypes may be considered as the two extremes of a unique evolving disease. Since the evolution of lung damage may eventually include a course leading to prevalent fibrosis [10], these structural and anatomic alterations, if present, result in significant changes of respiratory mechanics with substantial implications for the application of mechanical ventilation and ventilatory setting [11]. Therefore, considering the peculiar mechanical properties of a fibrotic lung, we here propose a further COVID-19-related phenotype, the “fibrotic” type F, showing either fibrotic appearance on CT scan, fragile lung mechanical features and functional derangement resulting from the static strain.
3. Molecular and Mechanical Mechanisms Driving COVID-19 Phenotype Transition
Progression from one phenotype to another may depend on the excessive activation of two main pathways: (1) Aggressive inflammatory response to SARS-CoV-2 infection; (2) physical mechanisms driven by the pulmonary stretch.
Virus infection of lung cells may cause a highly inflammatory form of programmed cell death (pyroptosis), with the secretion of cytokines and chemokines [12][13][12,13]. In individuals with the dysfunctional immune response, this process may result in a severe local and systemic inflammatory storm [14], activation of coagulation, and several procoagulant pathways (thrombo-inflammation or immune-thrombosis) [15]. So far, COVID-19-related endotheliitis and microcirculatory clot formation were reported in post-mortem studies [16][17][18][19][20][16–20]. Progression from type L to type H phenotype can be caused by both further mechanisms of inflammatory amplification overlapping the host inflammatory response phase and by excessive mechanical stress acting on the lung parenchyma sustained by self-inflicted lung injury (SILI) [17][18][19][17–19]. At this stage, in patients with phenotype H, the activation of multiple aberrant host pathways might result in impairment of the mechanisms of lung repair, promoting fibrotic changes and driving the progression towards type F [20]. This evolution may be forecasted through a structural change to the lung scaffolding associated with imbalance between profibrotic (TGF-α, TGF-β, interleukin-1β, platelet-derived growth factor) and antifibrotic (prostaglandin E2, keratinocyte growth factor, hepatocyte growth factor) mediators [21][22][21,22]. Indirect evidence in animals and experimental models suggest that both vascular lesions with chaotic repair and angiogenic responses [23] and biophysical insult driven by the mechanical ventilation itself or by the excessive activation of respiratory drive [24][25][24,25], could have a key role in this evolving process. In vitro and animal model studies show that mechanical stretch of lung epithelial cells results in TGF-α activation and the lung remodeling process after mechanical ventilation [25].
Figure 1 illustrates the peculiar pathophysiological changes of the lung and the “phenotypes progression” during severe SARS-CoV-2-related respiratory failure.
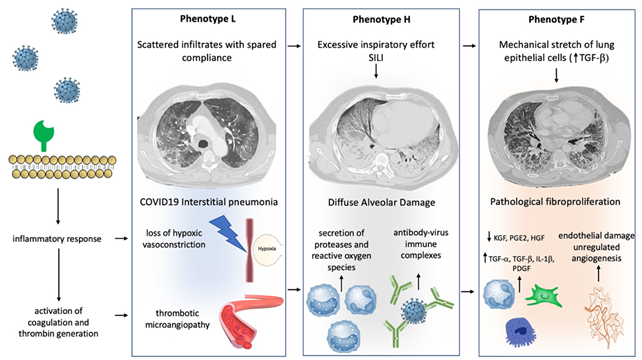
Figure 1. Mechanisms of progression between phenotype: Different hypothetical mechanisms, involving physical stimuli and biological modifications, can determine the progression between COVID-19 phenotypes. Progression from type L to type H may result from excessive inspiratory effort (SILI) and from recruitment of neutrophils into the lung parenchyma with secretion of proteases and reactive oxygen species. Furthermore, a role of activation of Fc receptors immune cells through antibody-virus immune complexes has been hypothesized. Progression from type H to type F results from damage to the scaffolding of the lung and vascular lesions with disorganized repair and imbalance between profibrotic and antifibrotic mediators. Physical factors, such as lung parenchyma stretch, may also contribute via transforming growth factor-beta (TGFB) secretion. See the text for more details.
4. The Role of Spontaneous Breathing in COVID-19-Related Lung Injury
In general, maintenance of spontaneous breathing in patients with ARF under ventilatory support has many positive effects such as: Improving oxygenation, preventing the mass loss and atrophy of the peripheral muscles, protecting against the diaphragm dysfunction, reducing the need for pharmacological sedation, and curbing the incidence of delirium [26][27][26,27]. Notwithstanding, the critical role of intense respiratory effort and high respiratory drive to foster the progression of lung damage and to favor a myo-diaphragmatic trauma has been demonstrated both in animal models and in humans [28][29][30][31] [28–31]. In patients with ARDS the inspiratory effort might be affected by different stimuli not always subjected to ventilatory support [32]. In particular different degrees of lung inflammation could influence respiratory drive irrespectively of gas exchange impairment [33]. In animal models of acute lung injury the inflammatory cascade seems to enhance inspiratory effort through the activation of pulmonary C-fibers, vagal-nerve stimulation, and pulmonary stretch receptors inhibition [34]. In patients with COVID-19, the direct invasion of respiratory centers due to SARS-CoV-2, may cause alteration of respiratory drive, thus affecting inspiratory effort [35].
A reliable method to assess changes in pleural space (Ppl) during spontaneous breathing is by using an esophageal balloon catheter to measure esophageal pressure (Pes) as a surrogate of Ppl [36]. A recent study estimated the intensity of spontaneous breathing effort by Pes measurement in 30 patients with hypoxemic ARF [29], and confirmed that vigorous effort is present as in typical ARDS patients [2]. In injured lungs, lung tissue becomes inhomogeneous as a consequence of inflammation and edema, and the distribution of the forces applied to the parenchyma during spontaneous breathing becomes asymmetrical (“solid-like” behavior, opposed to the “liquid-like” behavior typical of healthy lungs) [37][38][39][37–39]. In particular, the negative swing in pleural pressure generated by diaphragmatic contraction is not evenly transmitted and tends to concentrate in dependent regions near the diaphragmatic interface, where high local values of PL are developed. This impaired distribution of physical forces during spontaneous breathing causes a pendelluft phenomenon and can result in a local overstretch of the dependent lung [40][20,40]. In type F lung phenotype, lungs are a patchwork of different tissue elasticities, due to the contiguity of preserved lung tissue and areas of dense anelastic parenchyma [11]. During spontaneous breathing, Ppl swing distribution is even more inhomogeneous and unpredictable, lung tissue deformation occurs unevenly, and some lung areas can reach a harmful stress/strain level. These processes described in COVID-19 injured lungs can result in SILI with diffuse alveolar damage and could be therefore considered as main determinants for the lung phenotype progression over the course and the spectrum of disease severity (Figure 2). In this scenario, it should be noted that a relevant feature in patients experiencing COVID-19 pneumonia is the lack of perceived dyspnea despite severe hypoxemia (the so-called silent hypoxia) [41][42][43][41–43]. Recently, a study evaluated airway occlusion pressure (P01), a surrogate measure of respiratory drive, in mechanically ventilated COVID-19 patients. In this cohort of patients, P01 was frequently above 4 cmH2O, suggesting high neuronal respiratory drive, high respiratory effort, and excessive respiratory muscles load [44]. It has been described that COVID-19 patients can maintain a (pseudo)normal respiratory rate despite an increase in inspiratory effort, thus indicating that PL and inspiratory effort cannot be estimated by the individual’s breathing frequency [45][46][47][48][49][45–49]. Pes monitoring could help in the identification of patients with excessive inspiratory effort who are at risk of SILI and progression towards more serious lung phenotypes.
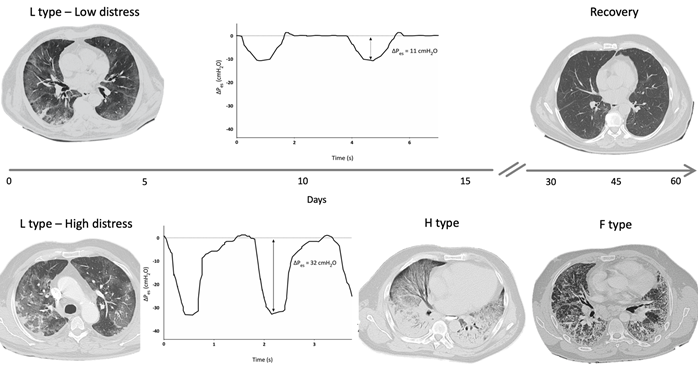
Figure 2. Hypothetical role of inspiratory effort in progression from type L to type H and F: Figure 2 shows DPes monitoring in two patients with SARS-CoV-2 pneumonia undergoing non-invasive ventilation (NIV) with Helmet. Patient 1, at the top of the figure, shows type L phenotype on computed tomography (CT) scan, and modest inspiratory effort (DPes 11 cmH2O). Over time, a progressive clinical and radiological improvement with ground glass changes resolutions was recorded. Patient 2, at the bottom of the figure, shows significant inspiratory effort (DPes 32 cmH2O). Over time, there was a progression from type L to type H required intubation and invasive mechanical ventilation. Progression from type H to type F was documented after 45 days from Intensive Care Unit admission. DPes: Change in esophageal pressure.