Sodium glucose co-transporter 2 (SGLT2) inhibitors are effective antihyperglycemic agents by inhibiting glucose reabsorption in the proximal tubule of the kidney.
- SGLT2 inhibitors
1. Introduction
There are six identified SGLT (sodium glucose co-transporter) proteins in humans, of which the SGLT1 and SGLT2 receptors have been studied more thoroughly in recent years. Despite the outstanding sequence similarity between SGLT1 and SGLT2, they show different physiological and biochemical properties. While SGLT1 is primarily expressed in the intestines, SGLT2 is most abundant in the renal cortex, where it plays an essential role in renal glucose reabsorption. SGLT2 inhibitors, including dapagliflozin, canagliflozin, ipragliflozin, tofogliflozin, luseogliflozin, sotagliflozin, ertugliflozin and empagliflozin, have been studied in several clinical studies for the treatment of type 2 diabetes mellitus. Their selectivity for the SGLT2 receptor shows significant variance. While empagliflozin is 2500×, ertugliflozin is 2000×, dapagliflozin is 1200×, canagliflozin is 250×, sotagliflozin is only 20× more selective for the SGLT2 receptor over SGLT1, which may cause significant differences in the mechanism of action [1]. Several studies have proved that SGLT2 inhibitors are associated with reduced cardiovascular morbidity and mortality, including heart failure, and vascular diseases [2][3][4][5][6]. The underlying hypothetic mechanisms of SGLT2 inhibitors beyond their antidiabetic effects are summarized in
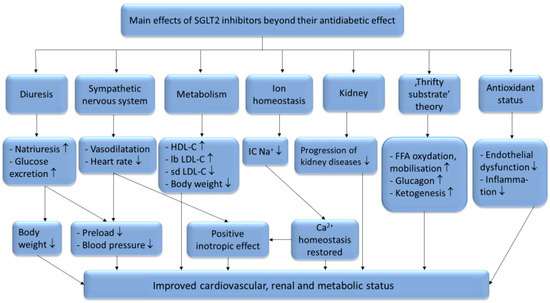
Figure 1.
+: intracellular sodium-ion; FFA: free fatty acids; ↑: increased amount; ↓: decreased amount.
Interestingly, these cardiovascular and renoprotective effects occur despite an increase in low-density lipoprotein cholesterol (LDL-C) levels which was observed in several clinical studies [11][12]. Nevertheless, this increase in LDL-C happens in the setting of other beneficial changes in plasma lipoprotein metabolism. In this review, we aim to analyze the effects of SGLT2 inhibitors on lipid metabolism to better understand their beneficial effects on cardiovascular outcomes despite slightly elevating LDL-C level.
2. The SGLT2 Inhibitors’ Effect on Plasma Lipoprotein Levels
Several studies reported lowered serum levels of total cholesterol (TC) and triglycerides (TG) [13][14][15] as a result of SGLT2 inhibitor therapy, however there is a debate regarding the changes observed in the serum levels of high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C). According to Calapkulu et al. LDL cholesterol level decreased by 13.4 mg/dL after 6 months in diabetic patients with dapagliflozin (10 mg/die) [13], In contrast, Cha et al. reported an increase of 1.3 mg/dL in LDL level after 24 weeks of dapagliflozin add-on therapy [16], in concordance with the results of Basu et al. in mice [17]. Furthermore, according to Schernthaner et al. canagliflozin (300 mg/die) caused 11.7% increase in LDL after 52 weeks of therapy in patients with type 2 diabetes mellitus [18]. According to Basu et al. the cause of this possible increase in the LDL-C levels could be due to an increased lipoprotein-lipase (LpL) activity and because of a delayed turnover of LDL in the circulation. Canagliflozin reduced the expression of angiopoetin-like protein 4 (ANGPTL4), which is a known inhibitor of LpL in white and brown adipose, skeletal muscle, and heart tissues. With greater LpL activity both the TG and the VLDL levels decreased. They also observed significantly delayed LDL turnover compared to the control group, which can originate from the lowered hepatic levels of the LDL-receptor, which is the major receptor for the clearance of plasma LDL [17]. Another important factor is the ratio of the different LDL subclasses. Using gradient gel electrophoresis (GGE) LDL particles are classified into 4 subclasses, including large (LDL I), intermediate (LDL II), small (LDL III), and very small (LDL IV) LDLs [19][20]. LDL I and II, also referred to as large buoyant (lb) LDL, and LDL III and IV as small dense (sd) LDL particles [21][22]. Small dense LDL particles are more prone to induce metabolic disorders [23][24], obesity [25][26], type 2 diabetes [20][27] and coronary artery disease (CAD) [28] due to their longer circulation time than that of large LDL particles [29], enhanced ability to penetrate the arterial wall and higher susceptibility to oxidation [30]. It is generally known that modified (oxidized and glycated) LDL particles are highly atherogenic and possess more proinflammatory properties than native LDL molecules [28][31]. Interestingly SGLT2 inhibitors slightly increase LDL level yet have beneficial effects on CV morbidity and mortality. The contradiction may be resolved by the results provided by Hayashi et al. showing that dapagliflozin decreased sd LDL, and increased lb LDL levels after 12 weeks of dapagliflozin therapy (5 mg/die) in type 2 diabetic patients [15]. This effect on LDL subclasses ratio may play a significant role in SGLT2 inhibitors’ cardioprotective property [32][33], provided it is a class effect.