Autophagy (self-eating) is a multifaceted and evolutionarily preserved sequence of actions that is activated in response to dysfunctional organelles and aggregated protein to sustain cellular homeostasis.
- autophagy
- glioblastoma
- glioma stem cells
- chemoresistance
1. Introduction
Autophagy (self-eating) is a multifaceted and evolutionarily preserved sequence of actions that is activated in response to dysfunctional organelles and aggregated protein to sustain cellular homeostasis [6][1]. The autophagy pathway commences by a series of main active proteins, such as UV radiation resistance associated gene (UVRAG), autophagy-related (ATG) proteins, phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), light chain 3 (LC3), Beclin 1 and ubiquitin-binding protein (p62) [7][2]. These proteins collectively lead to the development of double-membrane autophagosomes, and ultimately lead to the degradation of inner substances via fusion with lysosomes. Therefore, autophagy has been implicated in various diseases, including cancer [8][3]. Earlier, autophagy was assumed to be correlated to apoptosis [9][4]. Recently, a contradictory impact of autophagy, specifically the promotion of cell survival, has been widely studied [10][5]. This “double-edged sword” outcome (apoptosis + cell survival) in tumorigenesis varies depending upon the reaction of cells to precise stimulation and different cancer types. In the tumor microenvironment, elevated autophagy levels allow cancer cells to endure, recommencing initiation and proliferation [11][6].
2. Autophagy: The Process of Cellular Recycling
The word autophagy is originated from two Greek words, “auto” and “phagy”, which means self-eating [31][7]. Autophagy, a cell survival process, is responsible for maintaining cellular homeostasis under unusual stimuli like nutrient starvation, hypoxia, pathogen infection and oxidative stress. It is distinguished by the formation of the autophagosome, which is a double-membranous vesicle fusing with the lysosome to distribute the cytoplasmic contents for degradation. Autophagy also modulates cell health and longevity via “housekeeping” and protein quality control, which affect the regulation of ageing, immunity, neurodegeneration and cell death [32,33,34][8][9][10]. Additionally, autophagy increases in response to chemotherapeutic agents, thereby generating resistance and inhibiting the anticancer effect of drugs. Several examples showed that by hindering this pro-survival activity of autophagy using genetic or pharmacological agents, tumor cells could be targeted by inducing apoptosis. However, a continuous autophagic signal leads to autophagic cell death due to exhaustion of critical organelles and proteins. Therefore, autophagy plays a two-faced role in cancer, working either as a guardian or as a killer of cancer cells, depending upon the different cancer stages and the surrounding environment. Hence, targeting autophagy could be used as a promising approach for cancer treatment [35][11].
Depending on the selection and delivery of cargo to the lysosome, there are three different autophagy types, namely, macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Microautophagy is the only constitutive autophagy mechanism that causes engulfment of small cytoplasmic debris into the lysosome for degradation, whereas macroautophagy (the main form of autophagy) is developed in response to stress and targets the delivery and degradation of any kind of aggregated proteins and dysfunctional organelles by creating a special form of cytosolic vesicles, known as an autophagosome. CMA is also introduced in response to prolonged stress, which targets aggregated proteins with a pentapeptide motif KFRQ for degradation in the lysosome [36][12]. The different types of autophagy are shown in Figure 1.
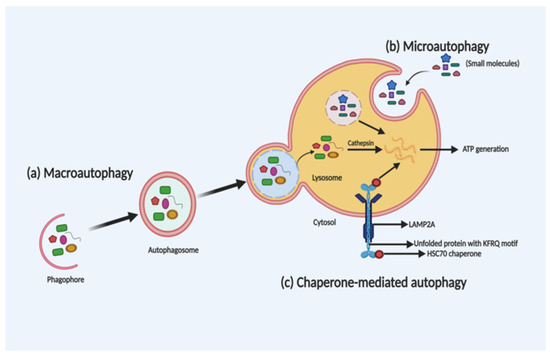
Figure 1. Schematic illustration of the different types of autophagy. Macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) are three different types of autophagy based on the selection of the cargo and delivery approach for degradation in the lysosome. In macroautophagy (a), autophagosome-mediated degradation takes place by engulfment of dysfunctional organelles and aggregated proteins. In microautophagy (b), direct engulfment of small molecules takes place via the lysosome for degradation. Whereas, in CMA (c), unfolded proteins with a KFRQ motif is degraded in the lysosomes through receptor-facilitated transfer.
Extensive research in genomics has led to the development of a new era in targeted cancer therapies that offer hope to cancer patients whose intensive chemotherapeutic treatment is improbable to give a positive result. The detection of the unique molecular characteristics in glioblastoma cells responsible for making them distinctive from the normal cell has led to the development of therapies that solely target these special kinds of genetic lesions, and overcoming chemotherapy-induced toxicity. However, the problem of resistance is found regardless of the target and the mechanism of action [37][13]. The presence of cell-mediated autophagy indicates a relationship between chaperones and autophagy, so it can be hypothesized that if there is any structural or functional abnormality or defect in the chaperones, it should also affect the autophagic process. Under adverse microenvironments and chemotherapy, autophagy plays a significant part in the tumor cells’ adaptation process and facilitates cell survival, which serves as a critical adaptive response during starvation and stress, leading to the recycling of energy and nutrients [38][14]. The dynamic role of autophagy is dependent upon the tumor development stages [39][15]. Autophagy may play a two-fold role in tumorigenesis by functioning equally as a tumor promoter as well as a tumor suppressor. Although autophagy may cause hindrance in cancer treatment at the late stage, it shows promise as a new target for the development of anticancer therapies for glioblastoma [40][16]. The influence of autophagy on tumorigenesis and treatment response gained attention after elucidation of the mechanisms behind autophagy.
3. Molecular Mechanism of Autophagy: Sliding on the Edges of the Sword
Under the starvation condition, the autophagic process is divided into five main steps: (1) nucleation; (2) elongation; (3) maturation; (4) fusion; and (5) degradation. Autophagosome formation is the hallmark of the beginning of autophagy. The target molecules are encapsulated by autophagosomes, which further interacts with the lysosomes. This interaction leads to the formation of autolysosomes, where the aggregated proteins or dysfunctional organelles undergo degradation. Phagophore formation is the early stage of autophagy, which is formed by the endoplasmic reticulum, Golgi complex, plasma membrane and mitochondrial membrane [41,42,43][17][18][19]. The phagophore extends and sequesters the cargo proteins for degradation to form the autophagosome, which further fuse with a lysosome to generate an autolysosome. Then, the autophagosomal contents get degraded by acidic hydrolases of the lysosomes in a pH-dependent manner [44][20]. The formed catabolic products are implemented in several metabolic processes and undergo degradation for generating adenosine triphosphate (ATP).
ATGs regulate autophagy and was discovered initially in yeast [45][21]. Over 35 different ATGs have been identified in yeast up to date, from which some of them have their homologues expressed in mammals [46,47][22][23]. Macromolecular complexes or protein groups, for instance, the autophagy-specific PI3K complex, the unc-51 like autophagy activating kinase 1 (Ulk1) and its regulators and the transmembrane protein Atg9, regulates the initiation of phagophore formation in mammals [48,49][24][25]. The PI3K complex (active enzyme VPS34, i.e., class III PI3K, a mammalian homolog of yeast Vps15 (p150), and Beclin 1 and Atg14) catalyzes the fabrication of phosphatidylinositol-3-phosphate. This leads to the assembly of effector proteins, like the WD-repeat domain phosphoinositide-interacting (WIPI) family proteins and double FYVE containing protein 1 (DFCP1) [50,51][26][27]. Two ubiquitin-like conjugation systems (Atg12–Atg5–Atg16L and phosphatidyl-ethanolamine (PE)-light chain 3, LC3) help in the elongation process of the isolation membrane and for the formation of the autophagosome. In the first system, i.e., the Atg12–Atg5–Atg16L complex, Atg12 binds to Atg5, forming the Atg5–Atg12 complex, with the assistance of Atg7 and Atg10 (E1 and E2). The Atg5–Atg12 complex further oligomerize and develops into a larger Atg16L complex through its interaction with the Atg16L, which is localized on the outer facade of the extended autophagosomal membrane. However, the Atg16L complex gets disconnected from the autophagosomal membrane forming the autophagosome [52][28]. The second system, namely, the LC3 system (mammalian homolog of yeast Atg8), which is cleaved by cysteine protease, Atg4, further conjugates to the lipid PE by the action of Atg3 (E2-like enzyme) and Atg7 [53][29]. LC3II (a lipidated form of LC3) supplies the growing autophagosomes to further recruit cargo adaptor proteins (autophagy receptors), such as neighbor of BRCA1 gene 1 (Nbr1), NIX or p62. Adaptor proteins promote the cargo’s recruitment, such as for the damaged organelles and ubiquitinated aggregates of proteins, from the cytoplasm to the autophagosome [54,55][30][31]. Post-completion of the autophagosome formation, it fuses with lysosomes via several unknown mechanisms in a mammalian cell. A few regulators, such as LC3 (the AAA-type ATPase SKD1) and Rab7 (the small GTP-binding protein), are involved in the process of autophagosome–lysosome fusion [56,57][32][33]. Autolysosome formation then finally activates the hydrolases and cargo degradation. The molecular mechanism of autophagy is illustrated in Figure 2.
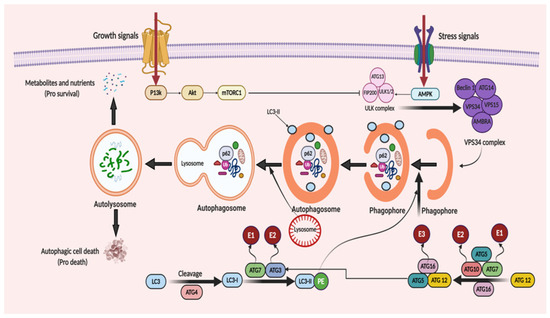
Figure 2. When cellular energy is sufficient, mammalian target of rapamycin complex 1 (mTORC1) becomes activated and phosphorylates each member of the ULK complex to prevent autophagy induction. In the case of glucose absence or cellular stress conditions, AMP-activated protein kinase (AMPK) becomes activated to initiate autophagic flux. This flux starts with the formation of the ULK complex consisting of ATG13, ULK1/2 and FIP200 (a ULK-interacting protein). The activity of mTORC1 is prohibited, non-phosphorylated ATG13, ULK1/2 and FIP200 forms a complex and, as a result, autophagy is started. The initiation of phagophore formation is achieved with the gathering of the ATG14, Beclin 1, VPS34, VPS15, and AMBRA proteins to generate the VPS34 complex. This also affects the further continuation of autophagic activity with downstream constituents. This takes place with the help of two ubiquitin-like conjugation systems. The first system comprises of ATG12 protein binding. Firstly, ATG7, which is linked to ubiquitin-activating enzyme E1, activates ATG12. Then, with the conjugation of ATG10 linked to ubiquitin-activating enzyme E2, ATG5 and ATG16, a huge protein complex is formed to contribute to the progression of the autophagosome. Similarly, in the second system, with the help of ATG4 protein, LC3 is modified to LC3-I. Then, LC3-I is attached to phosphatidylethanolamine (PE) to form LC3-II-PE catalyzed by ATG7 linked to E1, and ATG3 linked to E2. LC3-II-PE binds the membrane surface of the autophagosome and helps the attachment of adaptor proteins. When the phagophore formation is completed, it is named as a mature autophagosome. After fusion of the lysosome and autophagosome, an autolysosome is formed and it leads the degradation of the materials, recycles the nutrients and metabolites to supply energy (pro-survival) and triggers the autophagic cell death (pro-death).
3.1. Autophagy Promotes Tumor Progression in Gliomas
It has been revealed that stress-mediated autophagy induction in tumor cells can lead to resistance mechanisms against the treatments with subsequent tumor progression and recurrence [4]. Noor et al. suggested that in a KRAS-driven glioma mouse model, the autophagy inhibition considerably diminished the growth of glioma and oncogenic progression by genetically suppressing ULK1 or Atg7 and Atg13, showing that autophagy is vital during the early phase in glioma pathogenesis [58][34]. Autophagy can also be linked to glioma progression, specifically high-grade gliomas. The levels of LC3 and p62 are notably associated with a worse prognosis, implying that LC3 and p62 could be regarded as valuable prognostic aspects of glioma [59][35]. Furthermore, numerous glioblastoma patients have shown an overexpression of autophagy-associated proteins with amplification of ULK1/ULK2 and transcription factor EB (TFEB) [60][36]. Wen et al. observed that an elevated expression of the ATG4C transcript in high-grade glioma patients was associated with shorter overall survival (OS) time. In T98G glioma cells, ATG4C knock-down repressed autophagy and this facilitated the arrest of cell cycle and apoptosis promotion via reactive oxygen species (ROS) production, p21, TP53, BCL-2-associated X protein (Bax) expressions and decreased Bcl-2 levels [61][37]. Reduced ATG4C expression induced the sensitivity of the T98G and U87-MG glioma cells to TMZ by autophagy inhibition. Moreover, ATG4C knock-out (KO) appreciably reduced the growth rate of glioma in nude mice [61][37]. The quantification of p62, LC3B, Beclin-1 and BAG3 (autophagosomal molecules) proved that nutrient or oxygen deprivation enhances the autophagy in astrocytoma compared to normal brain tissue [62][38]. A long, noncoding RNA (Malat1) activates autophagy and endorses cell proliferation by inhibition of miR-101, which further decreases the expression of autophagy-associated genes such as ATG4D, Stathmin 1 (STMN1) and RAB5A (Ras-related protein Rab-5A) [63][39]. The Malat1 levels drastically increased in glioma biopsy samples with regard to flanking normal tissue [63][39]. Hypoxia (~3–0.1% oxygen) stimulates the hypoxia-inducible factor 1-alpha (HIF-1α) activation, thereby promoting autophagy via regulation of transcription of autophagic genes, such as coding for the Bcl-2/E1B 19-kDa-interacting protein (BNIP3) together with the BNIP3L, ATG5 and Beclin-1 gene (BECN1) [64,65][40][41]. BNIP3/BNIP3L stimulates autophagy via Beclin-1 release from the Bcl-2/Beclin-1 or Bcl-xL/Beclin-1 complexes [66][42]. Besides, HIF-1α stimulates angiogenesis to ensure the oxygen and nutrient availability for tumor cell survival via VEGF transcriptional regulation [67][43]. The expression of angiogenic and hypoxia levels is interconnected with tumor grade and a poor prognosis in brain tumor patients [68][44]. Hai-Bo et al. studied that amplification in the formation of vasculogenic mimicry (VM) is associated with a poor prognosis and an elevated expression of pKDR/VEGFR-2 and ATG5 in glioma patients. Autophagy can stimulate VM through pKDR/VEGFR activation by ROS generation and the subsequent activation of the PI3K–AKT pathway in glioma stem cells [69][45]. These outcomes support the key role of autophagy in the resistance and aggressiveness of hypoxic glioma regions, which further supports the survival of tumor cells. These outcomes also suggest that autophagy also plays a key role in the migration, proliferation and invasion of these tumor cells. Autophagy also supports cell growth in the tumor microenvironment. Additionally, in tumor cells, oxidative stress induces the activation of pro-autophagy factors, for instance, BNIP3L, LC3, BNIP3, ATG16L, HIF-1α and nuclear factor kappa B (NF-κB) to promote caveolin-1 (Cav-1) degradation, ultimately resulting in autophagy activation. Further, extended treatment with TMZ was found to induce glioma cell line dormancy. Histone cluster 1 H2b family member K (H2BK), ephrin type-A receptor 5 (EphA5) and the insulin-like growth factor binding protein 5 (IGFBP5) have been suggested to be related to the activation of the dormancy. Interestingly, it was found to be accompanied with acquired stemness, mediated via the expression of stem cell markers, including octamer-binding transcription factor 4 (OCT4), kruppel like factor 4 (KLF4), and sex-determining region Y-box 2 (SOX2) [70][46]. Thus, these tumor dormancy-regulating signaling pathways could be prospective therapeutic targets to holdup or impede glioblastoma recurrence after surgery [71][47].
3.2. Autophagy Suppresses Tumor Progression in Gliomas
Autophagy has been demonstrated to inhibit the tumor initiation stage, eliminating cancer cells during tumor progression. The deletion or lower expression of important genes (Beclin-1, FIP200, blood-inducing factor 1 (Bif1), UVRAG, Atg4c and Atg5) is reported in gliomas for autophagosome initiation and elongation [72][48]. Earlier findings have reported the lower levels of Beclin-1 transcript in glioblastoma [73][49]. Notably, the elevated levels of Beclin-1 and LC3 were linked to better survival in glioma patients [74,75][50][51]. Higher AKT and mTOR hyperphosphorylation (activation) has been reported in high-grade gliomas (Grade III and IV) as compared to low-grade ones (Grade I and II) [76,77][52][53]. Additionally, the mTOR signaling pathway activation associates with the autophagy inhibition, supporting the glioma stem cell proliferation and pluripotency [78][54]. Likewise, glioma stem cells endorsed tumor infiltration, therapeutic resistance and malfunction of treatment [79][55]. MiR-224-3p suppresses ATG5 and FIP200, thereby inhibiting autophagy, and its overexpression restrained the tumorigenesis in glioblastoma cells [80][56].
The upregulation of BNIP3 (pro-cell death Bcl-2 family member) in hypoxia induces autophagy in glioma cell lines [81][57]. Moreover, autophagy may restrain tumorigenesis by the removal of the p62-tagged aggregates. The accumulation of p62 leads to damage of the proteins, DNA and mitochondria, as well as ROS generation, thereby promoting an unstable genome and tumor progression [82][58]. P62 overexpression is linked to poor prognosis in glioblastoma patients [75][51]. Jiang et al. showed that sinomenine hydrochloride (SE) induces autophagic cell death in glioma cells via ROS generation, consequently activating the c-Jun N-terminal kinase (JNK) pathway and inhibiting the AKT/mTOR pathway [83][59]. Autophagy mediates senescence; hence, it inhibits the malignant transformation as well [84][60]. Yuan et al. also confirmed that resveratrol improved the TMZ toxicity with increasing ROS production, which stimulates the 5′ AMP-activated protein kinase (AMPK) activation and the subsequent inhibition of the mTOR pathway, as well as decreases the Bcl-2 level [85][61]. Flovokawain (chalcone) inhibits cell proliferation in the U87, T98 and U251 glioma cell lines via activating autophagy and subsequent senescence mediated by ER stress; moreover, it also inactivated the AKT/mTOR pathway [86][62]. Autophagy may also interrupt the formation/progression of tumor through ATG protein-mediated induction of apoptosis [87,88][63][64]. It has been proposed that Beclin-1 might exert its pro-apoptotic effect with preventing the anti-apoptotic role of Bcl-xL and Bcl-2. Huang et al. suggested that Beclin-1 induced apoptosis via Bcl-xL and Bcl-2 binding, consequently releasing BCL2-antagonist/killer (Bak) and Bax, which further activates caspases-3/-9 in glioma cells [89][65].
3.3. The Bipolar Role of Autophagy
As discussed in the previous section, autophagy has been implicated in cellular defense and the oncogenic process. Although the exact mechanism of this shift is not exactly understood, it can be linked to the crosstalk between cellular apoptosis and autophagy [90,91][66][67]. Autophagy removes damaged organelles and proteins in the cell and protect the cell from their deleterious effect. In contrast, autophagy activated in response to cellular stress promotes cell survival in case of defective apoptosis. Apoptosis is a form of programmed cell death. Since autophagy and apoptosis both play crucial roles in the homeostasis of cells, the crossover of these two pathways is not surprising. These two signaling pathways cross each other at several nodes, involving interactions between (1) ATG12 and Mcl-1; (2) UVRAG and BAX; (3) Beclin1 and BCL-2; (4) Caspase and Beclin1; (5) ATG12 and ATG3; (6) ATG5 and FADD; and (7) P53 cross regulation [92,93,94,95,96,97,98,99][68][69][70][71][72][73][74][75]. These crossover nodes provide an opportunity for further investigation and also therapeutic interventions.
References
- Ryter, S.W.; Cloonan, S.M.; Choi, A.M. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol. Cells 2013, 36, 7–16.
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473.
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075.
- Berry, D.L.; Baehrecke, E.H. Autophagy functions in programmed cell death. Autophagy 2008, 4, 359–360.
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer–Issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358.
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967.
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.-U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975.
- Tang, Y.; Jacobi, A.; Vater, C.; Zou, L.; Zou, X.; Stiehler, M. Icariin promotes angiogenic differentiation and prevents oxida-tive stress‐induced autophagy in endothelial progenitor cells. Stem Cells 2015, 33, 1863–1877.
- Gatica, D.; Chiong, M.; Lavandero, S.; Klionsky, D.J. Molecular mechanisms of autophagy in the cardiovascular system. Circ. Res. 2015, 116, 456–467.
- Su, Z.; Wang, T.; Zhu, H.; Zhang, P.; Han, R.; Liu, Y.; Ni, P.; Shen, H.; Xu, W.; Xu, H. HMGB1 modulates Lewis cell autoph-agy and promotes cell survival via RAGE-HMGB1-Erk1/2 positive feedback during nutrient depletion. Immunobiology 2015, 220, 539–544.
- Pan, H.; Wang, Z.; Jiang, L.; Sui, X.; You, L.; Shou, J.; Jing, Z.; Xie, J.; Ge, W.; Cai, X. Autophagy inhibition sensitizes hepato-cellular carcinoma to the multikinase inhibitor linifanib. Sci. Rep. 2014, 4, 6683.
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417.
- Masui, K.; Gini, B.; Wykosky, J.; Zanca, C.; Mischel, P.S.; Furnari, F.B.; Cavenee, W.K. A tale of two approaches: Comple-mentary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcino-genesis 2013, 34, 725–738.
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734.
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010.
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y. Pro-motion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820.
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437.
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy 2010, 6, 301–303.
- Burman, C.; Ktistakis, N.T. Autophagosome formation in mammalian cells. Semin. Immunopathol. 2010, 32, 397–413.
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348.
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822.
- Klionsky, D.J. Regulated self-cannibalism. Nature 2004, 431, 31–32.
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42.
- Chan, E.Y.; Kir, S.; Tooze, S.A. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 2007, 282, 25464–25474.
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900.
- O’ Farrell, F.; Rusten, T.E.; Stenmark, H. Phosphoinositide 3‐kinases as accelerators and brakes of autophagy. FEBS J. 2013, 280, 6322–6337.
- Proikas‐Cezanne, T.; Robenek, H. Freeze‐fracture replica immunolabelling reveals human WIPI‐1 and WIPI‐2 as membrane proteins of autophagosomes. J. Cell. Mol. Med. 2011, 15, 2007–2010.
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336.
- Lane, J.D.; Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50.
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145.
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in au-tophagic maturation of erythroid cells. Nature 2008, 454, 232–235.
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358.
- Nara, A.; Mizushima, N.; Yamamoto, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 2002, 27, 29–37.
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439.
- Tamrakar, S.; Yashiro, M.; Kawashima, T.; Uda, T.; Terakawa, Y.; Kuwae, Y.; Ohsawa, M.; Ohata, K. Clinicopathological Significance of Autophagy-related Proteins and its Association With Genetic Alterations in Gliomas. Anticancer Res. 2019, 39, 1233–1242.
- Giatromanolaki, A.; Sivridis, E.; Mitrakas, A.; Kalamida, D.; Zois, C.E.; Haider, S.; Piperidou, C.; Pappa, A.; Gatter, K.C.; Harris, A.L. Autophagy and lysosomal related protein expression patterns in human glioblastoma. Cancer Biol. Ther. 2014, 15, 1468–1478.
- Wen, Z.-P.; Zeng, W.-J.; Chen, Y.-H.; Li, H.; Wang, J.-Y.; Cheng, Q.; Yu, J.; Zhou, H.-H.; Liu, Z.-Z.; Xiao, J.; et al. Knockdown ATG4C inhibits gliomas progression and promotes temozolomide chemosensitivity by suppressing autophagic flux. J. Exp. Clin. Cancer Res. 2019, 38, 298.
- Jennewein, L.; Ronellenfitsch, M.W.; Antonietti, P.; Ilina, E.I.; Jung, J.; Stadel, D.; Flohr, L.-M.; Zinke, J.; von Renesse, J.; Drott, U.; et al. Diagnostic and clinical relevance of the autophago-lysosomal network in human gliomas. Oncotarget 2016, 7, 20016–20032.
- Fu, Z.; Luo, W.; Wang, J.; Peng, T.; Sun, G.; Shi, J.; Li, Z.; Zhang, B. Malat1 activates autophagy and promotes cell prolifera-tion by sponging miR-101 and upregulating STMN1, RAB5A and ATG4D expression in glioma. Biochem. Biophys. Res. Com-mun. 2017, 492, 480–486.
- Mazure, N. M.; Pouysségur, J., Hypoxia-induced autophagy: cell death or cell survival? Current opinion in cell biology 2010, 22, (2), 177-180.
- De Francesco, E. M.; Pellegrino, M.; Santolla, M. F.; Lappano, R.; Ricchio, E.; Abonante, S.; Maggiolini, M., GPER mediates activation of HIF1α/VEGF signaling by estrogens. Cancer research 2014, 74, (15), 4053-4064.
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581.
- Jawhari, S.; Ratinaud, M.-H.; Verdier, M., Glioblastoma, hypoxia and autophagy: a survival-prone ‘menage-a-trois’. Cell death & disease 2016, 7, (10), e2434-e2434.
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progres-sion and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426.
- Wu, H.B.; Yang, S.; Weng, H.Y.; Chen, Q.; Zhao, X.L.; Fu, W.J.; Niu, Q.; Ping, Y.F.; Wang, J.M.; Zhang, X.; et al. Autopha-gy-induced KDR/VEGFR-2 activation promotes the formation of vasculogenic mimicry by glioma stem cells. Autophagy 2017, 13, 1528–1542.
- Lamoral-Theys, D.; Le Mercier, M.; Le Calvé, B.; Rynkowski, M.A.; Bruyère, C.; Decaestecker, C.; Haibe-Kains, B.; Bontempi, G.; Dubois, J.; Lefranc, F.; et al. Long-term temozolomide treatment induces marked amino metabolism modifications and an increase in TMZ sensitivity in Hs683 oligodendroglioma cells. Neoplasia 2010, 12, 69–79.
- Tiram, G.; Ferber, S.; Ofek, P.; Eldar-Boock, A.; Ben-Shushan, D.; Yeini, E.; Krivitsky, A.; Blatt, R.; Almog, N.; Henkin, J.; et al. Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J. 2018, 32, 5835–5850.
- Shukla, S.; Patric, I.R.P.; Patil, V.; Shwetha, S.D.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Santosh, V.; So-masundaram, K. Methylation silencing of ULK2, an autophagy gene, is essential for astrocyte transformation and tumor growth. J. Biol. Chem. 2014, 289, 22306–22318.
- Miracco, C.; Cosci, E.; Oliveri, G.; Luzi, P.; Pacenti, L.; Monciatti, I.; Mannucci, S.; De Nisi, M.C.; Toscano, M.; Malagnino, V. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int. J. Oncol. 2007, 30, 429–436.
- Aoki, H.; Kondo, Y.; Aldape, K.; Yamamoto, A.; Iwado, E.; Yokoyama, T.; Hollingsworth, E.F.; Kobayashi, R.; Hess, K.; Shi-nojima, N. Monitoring autophagy in glioblastoma with antibody against isoform B of human microtubule-associated protein 1 light chain 3. Autophagy 2008, 4, 467–475.
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013, 32, 699–712.
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018, doi:10.1155/2018/9230479.
- Li, X.-Y.; Zhang, L.-Q.; Zhang, X.-G.; Li, X.; Ren, Y.-B.; Ma, X.-Y.; Li, X.-G.; Wang, L.-X. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J. Neuro-Oncol. 2011, 103, 453–458.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760.
- Jhanwar-Uniyal, M.; Jeevan, D.; Neil, J.; Shannon, C.; Albert, L.; Murali, R. Deconstructing mTOR complexes in regulation of Glioblastoma Multiforme and its stem cells. Adv. Biol. Regul. 2013, 53, 202–210.
- Guo, X.; Xue, H.; Guo, X.; Gao, X.; Xu, S.; Yan, S.; Han, X.; Li, T.; Shen, J.; Li, G. MiR224-3p inhibits hypoxia-induced au-tophagy by targeting autophagy-related genes in human glioblastoma cells. Oncotarget 2015, 6, 41620–41637.
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204.
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075.
- Jiang, Y.; Jiao, Y.; Wang, Z.; Li, T.; Liu, Y.; Li, Y.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits Human Glioblasto-ma Cell Growth through Reactive Oxygen Species Generation and Autophagy-Lysosome Pathway Activation: An In Vitro and In Vivo Study. Int. J. Mol. Sci. 2017, 18, 1945.
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803.
- Yuan, Y.; Xue, X.; Guo, R.B.; Sun, X.L.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546.
- Wang, J.; Qi, Q.; Zhou, W.; Feng, Z.; Huang, B.; Chen, A.; Zhang, D.; Li, W.; Zhang, Q.; Jiang, Z.; et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy 2018, 14, 2007–2022.
- Lépine, S.; Allegood, J.C.; Edmonds, Y.; Milstien, S.; Spiegel, S. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J. Biol. Chem. 2011, 286, 44380–44390.
- Zhu, Y.; Zhao, L.; Liu, L.; Gao, P.; Tian, W.; Wang, X.; Jin, H.; Xu, H.; Chen, Q. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell 2010, 1, 468–477.
- Huang, X.; Qi, Q.; Hua, X.; Li, X.; Zhang, W.; Sun, H.; Li, S.; Wang, X.; Li, B. Beclin 1, an autophagy-related gene, augments apoptosis in U87 glioblastoma cells. Oncol. Rep. 2014, 31, 1761–1767.
- Feng, F.; Zhang, M.; Yang, C.; Heng, X.; Wu, X. The dual roles of autophagy in gliomagenesis and clinical therapy strategies based on autophagic regulation mechanisms. Biomed. Pharmacother. 2019, 120, 109441.
- Su, M.; Mei, Y.; Sinha, S. Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 2013, doi:10.1155/2013/102735.
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against Fatal Sindbis Virus Encephalitis by Beclin, a Novel Bcl-2-Interacting Protein. J.Virol. 1998, 72, 8586–8596.
- Rohn, T.T.; Wirawan, E.; Brown, R.J.; Harris, J.R.; Masliah, E.; Vandenabeele, P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol. Dis. 2011, 43, 68–78.
- Yin, X.; Cao, L.; Peng, Y.; Tan, Y.; Xie, M.; Kang, R.; Livesey, K.M.; Tang, D. A critical role for UVRAG in apoptosis. Autoph-agy 2011, 7, 1242–1244.
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600.
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709.
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729.
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134.
- Feng, Q.; Zhang, Y.; Li, Y.; Liu, Z.; Zuo, J.; Fang, F. Two domains are critical for the nuclear localization of soluble adenylyl cyclase. Biochimie 2006, 88, 319–328.