Monoclonal antibodies are key therapeutic agents for several neurological conditions with diverse pathophysiological mechanisms, including multiple sclerosis, migraines and neuromuscular disease. In addition, a great number of monoclonal antibodies against several targets are being investigated for many more neurological diseases, which reflects our advances in understanding the pathogenesis of these diseases. Untangling the molecular mechanisms of disease allows monoclonal antibodies to block disease pathways accurately and efficiently with exceptional target specificity, minimizing non-specific effects. On the other hand, accumulating experience shows that monoclonal antibodies may carry class-specific and target-associated risks.
- monoclonal antibodies
- multiple sclerosis
- migraine
- neuromyelitis optica spectrum disorder
- myasthenia gravis
- Alzheimer’s disease
- inflammatory myopathies
- immune-mediated peripheral neuropathies
- Parkinson’s disease
- neurooncology
- Duchene’s muscular dystr
1. Introduction
The production of monoclonal antibodies (mAbs) was first described in 1975 when Köhler and Milstein developed methods for their isolation from hybridoma cells [1]. The ability to generate mAbs revolutionized antibody research and paved the way for tremendous clinical advances. For their discovery, Milstein and Köhler shared the 1984 Nobel Prize for Medicine or Physiology together with Niels K. Jerne for “theories concerning the specificity in development and control of the immune system and discovery of the principle for production of monoclonal antibodies”. According to the classical hybridoma method, mice were immunized with a mixture of antigens, their antibody-producing splenic B cells were fused with immortalized neoplastic B cells (myeloma cells) bearing a selection marker and the fused cells (hybridoma cells) were cultured in a selective medium. When visible colonies grew, their supernatants were screened for antibody production. For the first time, unlimited amounts of monoclonal antibodies specific for a single determinant could thus be produced in vitro. Köhler and Milstein did not patent their method, which facilitated the use of hybridoma technology by academics and the pharmaceutical industry for the generation of future potential therapies. At first, myeloma cells which retained the capacity to secrete their own immunoglobulin products were used. Later, such fusion was replaced by myeloma variants that express only one endogenous chain so that the fused cells secreted primarily or exclusively the antibody of the desired specificity. Besides their huge impact on research and diagnostic applications including epitope-specific immunoblotting, immunofluorescence, and immunohistochemistry, mAbs played an important role in therapeutics, contributing to the treatment of cancer, autoimmune and infectious diseases.
The first mAb approved by FDA for human use was a murine anti-CD3 monoclonal antibody, muromonab (OKT3), used for the treatment of organ transplant rejection [2]. However, murine mAb-associated allergic reactions (immune reaction against proteins from different species) led to the development of chimeric antibodies in 1984 [3]. Chimeric mouse-human antibodies were produced by grafting the entire antigen-specific domain of a mouse antibody onto the constant domains of a human antibody using recombinant DNA techniques [3]. Rituximab, a mouse-human chimeric mAb against the B-cell lineage marker CD20 was the first to be approved in 1997 by FDA for the treatment of relapsed or refractory, CD20-positive, B-cell, low-grade or follicular non-Hodgkin’s lymphoma [4]. Humanization of murine mAbs was achieved in the second half of the 1980s using CDR grafting methodology [5]. Later, the development of fully human monoclonal antibodies, in which both the variable region (Fab) and the constant region (Fc) are 100% human, was made possible through the advent of in vitro phage display technology and the generation of different mouse strains expressing human variable domains. Advanced antibody engineering technologies, such as phage display, affinity maturation, single B cell antibody technology and human antibody mouse are described in detail by Lu et al. [6]. The development of biosimilar mAbs has in many cases decreased the cost of treatment.
2. Basic Categories of Monoclonal Antibodies
2.1. Murine Antibodies
Murine antibodies are produced entirely from mouse protein and are the earliest mAbs developed. Due to the source of their production, they were recognized as allogeneic proteins, thus leading to polyclonal human anti-mouse antibody (HAMA) reactions, usually 2–3 weeks after their initial infusion [143][7]. HAMAs frequently had neutralizing action leading to rapid murine antibody inactivation or affected their pharmacokinetics promoting accelerated plasma elimination [144,145][8][9]. No murine mAb is currently in use in neurology.
2.2. Chimeric Antibodies
The serious limitations murine antibodies impose upon their clinical use, necessitated the development of new products with human components. Initially, the Fc portion of the antibody molecule, which dictates the functions of the antibody, was chemically exchanged with a human constant portion [146][10], giving rise to chimeric monoclonal antibodies. Chimeric mAbs contain 34% mouse protein in the variable region of the antibody, thus leading to a lower incidence of HAMA reactions compared to murine mAbs. Moreover, chimeric mAbs have a wide range of antigen specificities, increased cellular toxicity, and a beneficial pharmacokinetic and pharmacodynamic profile (longer half-life and increased affinity for the antigen) [147][11]. Rituximab and infliximab are the only chimeric mAbs currently in use in neurology (Table 1).
2.3. Humanized Antibodies
Advances in methods of molecular biology led to the development of humanized mAbs, which are 90% human, and only 10% mouse protein. Humanized mAbs are even less immunogenic compared to chimeric mAbs. Molecular techniques were used to further eliminate regions in the murine immunoglobulin chains that are not involved in the binding of antigen and to replace them with the corresponding human sequences. Complementarity-determining regions (CDRs) within the variable regions of both the heavy and light chains are of great importance in the binding specificity of the antibody. DNA fragments that correspond to the CDRs were grafted into the framework of human immunoglobulin genes using molecular methods [5]. Furthermore, replacement of some amino acid residues in the constant regions with the corresponding amino acids of the mouse “parental” monoclonal antibody proved advantageous [148][12]. Humanized antibodies retain the specificity and binding affinity of the “parental” murine mAbs, while being less immunogenic and acquiring biological functions of choice [149][13]. The great majority of mAbs in use or in development for neurological indications are humanized mAbs (Table 1 and Table 2).
2.4. Fully Human Monoclonal Antibodies
Peripheral blood lymphocytes or single cells derived from naïve and immunized donors were used to isolate immunoglobulin genes and to prepare libraries of plasmids with the cDNA’s of heavy and light chains. The combinatorial libraries were used to transfect bacteria which, in turn, were seeded on appropriate drug-supplemented agar medium Colonies producing active antibodies were then detected and isolated [150][14]. Phage display and transgenic mice technologies made production of 100% human mAbs possible [6]. Complete removal of murine components led to the production of mAbs that were mostly less immunogenic and, in many cases, improved their pharmacokinetic profiles slowing their clearance from plasma [147][11]. Erenumab and ofatumumab are fully human mAbs currently indicated for migraine prophylaxis and multiple sclerosis, respectively (Table 1). The human and murine components of murine, chimeric, humanized and human mAbs is schematically presented in Figure 1.
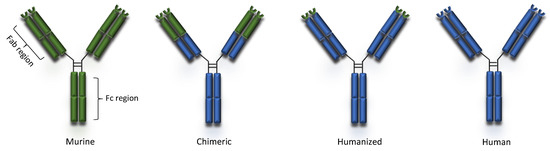
Figure 1. Types of monoclonal antibodies.
3. Mechanism of Action
Mabs may act through several direct and indirect mechanisms and some MAbs confer multiple mechanisms of action on a target [151][15].
3.1. Direct Mechanisms
Direct actions include antagonism of a soluble ligand or receptor, blockade of cell–cell interaction, agonism on a surface receptor activating certain signaling pathways within the target cell or inducing cell death [152,153][16][17]. The simplest form of antibody activity occurs when the antibody binds a soluble ligand, a cell-bound ligand, or a cell receptor, and blocks the binding of the ligand to the receptor, thereby disrupting the downstream signaling mediated by that receptor–ligand interaction. Examples of this activity is the binding of fremanezumab, galcanezumab and eptinezumab to the calcitonin gene-related peptide (CGRP) preventing it from signaling through the CGRP and Amylin-1 receptors [154,155][18][19].
Another approach is binding to a cell receptor in a non-agonistic manner to block ligand binding and activation of downstream signaling pathways as in the case of erenumab, which is an anti-CGRP receptor mAb [155][19]. Finally, cell–cell interactions between a cell-bound ligand and a cell-bound receptor on another cell can be blocked by mAbs, as in the case of natalizumab blocking lymphocytic transendothelial migration by binding to lymphocytic VLA-4 (CD49d) and preventing its binding to endothelial vascular cell adhesion molecule (VCAM) [55][20].
Agonistic mAbs mimic the activity of the normal ligand [151,156][15][21]. The agonist activity can occur when the antibody binds the receptor in a manner that mimics the binding of the natural ligand, resulting in antibody-mediated downstream signaling [156][21]. Alternatively, mAbs exerting agonist activity on receptors such as the tumor necrosis factor related apoptosis-inducing ligand (TRAIL) receptors initiate programmed cell death [157][22].
3.2. Indirect or Immune-Mediated Actions
Conserved differences in the constant regions (Fc) of IgG antibodies distinguish them into four subclasses: IgG1, IgG2, IgG3, and IgG4 [158,159][23][24]. These Fc regions are involved in binding to Fc receptors (FcγR), complement factor component 1q (C1q) and the neonatal receptor (FcRn) and as a result they determine the ability of different IgG subclasses to mediate effector functions such as phagocytosis, antibody-dependent cell-mediated cytotoxicity, complement activation and determine their half-life and capacity for transplacental transport and transport through mucosal surfaces [24] [159] Most unconjugated antibodies bear a human IgG1 Fc, an isotype that efficiently activates the immune system, with the scope of harnessing different immune cells and molecules towards target cell killing. Thus, IgG1 mAbs may activate natural killer (NK) cells through CD16A, induce antibody-dependent cytotoxicity (ADCC), bind to macrophage CD16A, CD32A and CD64 to promote antibody-dependent phagocytosis (ADPh) and activate the complement leading to complement-dependent cytotoxicity (CDC) [158][23]. More specifically, to trigger ADCC, the Fc binding domain of an antibody binds to a specific antigen expressed on the surface of a target cell. The antibody is then able to recruit NK cells to lyse the target cell [150][14]. CDC is triggered when the C1 complement factor binds an IgG1 or IgG3 antibody–antigen complex, resulting in the activation of the complement cascade culminating in the formation of the C5b-9 membrane attack complex (MAC) forming a water pore in the target cell leading to its lysis [160][25]. Most of the marketed mAbs such as alemtuzumab and rituximab belong to the IgG1 subclass and are shown to trigger ADCC and CDC [73,161][26][27]. The immune mediated mode of action of mAbs is schematically presented in Figure 2. On the other hand, IgG2 and IgG4 subclasses exhibit a lower affinity to the Fcγ receptor and are commonly preferred for blocking antigen function. More specifically, the IgG2 subclass is commonly selected to neutralize soluble antigens without inducing host effector mechanisms as in the case of erenumab and fremanezumab [154,155][18][19]. Similarly, IgG4 such as natalizumab and galcanezumab represent an important subclass of mAbs commonly selected when the recruitment of the host effector mechanisms is not desirable [55,155,159,162][20][19][24][28].
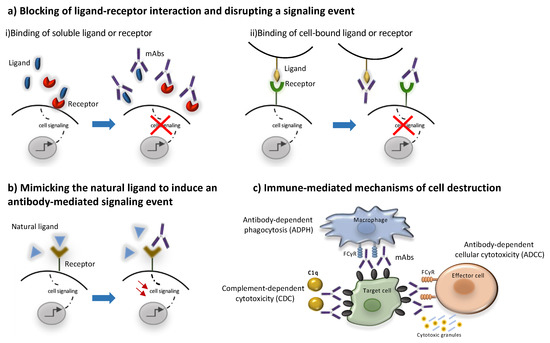
Figure 2. Mechanisms of action of monoclonal antibodies. MAbs may act through direct (a,b) or indirect mechanisms (c). The direct mechanisms include: (a) blocking ligand-receptor interactions through binding to (i) a soluble ligand or receptor or (ii) to a cell-bound ligand or receptor leading to inhibition of downstream signaling events, (b) agonism through binding to a receptor by mimicking its natural ligand leading to the activation of signaling pathways. Indirect mechanisms are immune -mediated as they involve the activation of certain types of immune cells and molecules to kill target cells (c). Most mAbs bear a human IgG1 Fc region that can activate effector cells, such as natural killer (NK) cells to induce antibody-dependent immune cell cytotoxicity (ADCC), or macrophage inducing antibody-dependent phagocytosis (ADPH), through the interaction with their FCγ receptors. Moreover, the Fc region of mAbs can activate the complement leading to complement-dependent cytotoxicity (CDC).
3.3. Conjugated mAbs
Conjugated mAbs are combined with a drug or a radioactive substance. These mAbs are currently used in oncology to deliver these substances directly to cancer cells [163][29]. They are specifically designed to induce either a block in proliferation or direct cell death (usually apoptosis) and can deliver higher concentrations of cytotoxic agents directly to the target cells without affecting normal cells, thus reducing the potential of adverse reactions [158][23]. Ibritumomab tiuxetan is an example of a radiolabeled mAb against CD20, (a B cell surface protein), which is conjugated with radioactive Yttrium-90 and used in radioimmunotherapy and Ado-trastuzumab emtansine (also called TDM-1), is an antibody that targets the HER2 protein conjugated to a chemotherapeutic drug called DM1 [164,165][30][31]. Although conjugated mAbs have neither clinical nor experimental application in neurology, they could be used in the future to destroy targets or traffic medications to specific cell types.
3.4. Bispecific Monoclonal Antibodies
Bispecific mAbs are especially designed to recognize and bind to two epitopes simultaneously. Their unique structure confers them an unlimited potential of novel functions. Combining the two distinct binding sites in a single molecule yields a compound function that is restricted both in space and time, which cannot be achieved by the administration of a mixture of two separate mAbs with the same specificity. Bispecific Abs can direct effectors cells to target cells, promote receptor internalization, deliver ligands to specific cell populations, simultaneously block two pathways or promote shuttling across biological barriers [166][32]. The latter is particularly relevant to neurology where the blood-barrier barrier (BBB) is an obstacle for access of mAbs to the CNS. One specificity of a bispecific Abs can be used to shuttle it through the BBB (e.g., binding to the transferrin receptor) and the second specificity can bind to protein targets to block or promote a process or destroy brain tumor cells [167][33].
Two bispecific Abs are currently marketed and many other are in development. As an example blinatumomab, which is indicated for Philadelphia chromosome-negative relapsed or refractory acute lymphoblastic leukemia binds simultaneously to the CD3 protein of T cells and to the CD19 protein of target neoplastic B cells. By binding to both proteins, it brings T effector cells in close proximity to target neoplastic cells promoting their immune-mediated lysis [168][34]. Emicizumab is another bispecific Ab approved in EU and US for Hemophilia A as it binds simultaneously coagulation factors IXa and X [169][35]. Many more other bispecific Abs are in clinical development for several uses [168][34]. No bispecific Abs are currently in use in neurological therapeutics. However, preclinical evidence hold promise for their use in neurology in the future. Delivery of the construct of a bispecific Ab with an LDLR-binding domain of apoB to facilitate its transfer across the BBB and promoting alpha secretase activity over beta-secretase activity thus favoring the neuroprotective APP cleavage by alpha-secretase using an adenoviral vector has shown beneficial effects in a mouse model of AD [170][36]. In addition, targeting simultaneously the angiogenic factor angiopoietin-2 (Ang-2) and translocator protein (TSPO), both of which are overexpressed in bevacizumab-treated glioblastomas, with a bispecific Ab in bevacizumab-treated rats resulted in prolonged survival [171][37]. Furthermore, another bispecific Ab targeting Ang-2 and vascular endothelial growth factor (VEGF) was also found to prolong survival in a mouse model with glioblastoma xenografts, suggesting that bispecific Abs targeting appropriate epitopes may be beneficial in neurooncology [172][38].
4. Doses, Routes of Administration and Pharmacokinetics
Regarding dosing, some mAbs are given in a fixed dose whilst others are given according to patient’s bodyweight. MAbs require parenteral administration for adequate bioavailability. In most cases mAbs are administered either intravenously (e.g., natalizumab) or subcutaneously (e.g., eremumab). Some can be administered by either route (e.g., rituximab), whilst intramuscular administration has also been reported (e.g., palivizumab). Intravenous administration is chosen for greater and faster bioavailability and lower risk of immunogenicity whilst subcutaneous use is chosen to avoid intravenous access and facilitate self-administration [147,173][11][39]. Subcutaneously administered antibodies are taken up by lymphatics and their plasma concentration increase slowly over several days. Circulating mAbs leave the vasculature by hydrostatic and osmotic pressure gradients. Their affinity for the epitope of their specificity determines their retention in target tissues [173][39].
The half-lives of mAbs vary from hours to several weeks [174][40]. MAb half-life is largely determined by the binding of the constant fragment (Fc) of humanized and human Abs of immunoglobulin G (IgG) class to the neonatal receptor FcRn, expressed on many adult cell types [147][11]. More specifically, IgG antibodies are thought to be taken up by catabolic cells by fluid-phase endocytosis. Although, under neutral pH, FcRn has a low affinity for IgG, the endosome content is then acidified, thus increasing the affinity of the FcRn for IgG. The FcRn-IgG complex is then re-shuttled to the cell surface where the IgG is released under neutral pH [175][41]. Proteins and antibodies in the endosome that are not bound to the FcRn undergo proteolysis. This is a salvage pathway recycling and protecting IgGs from degradation therefore increasing their half-life without affecting their function. The half-life of IgG1, IgG2 and IgG4 is in the range of 18 to 21 days whereas the half-life of other proteins with comparable molecular weight is significantly shorter. The half-life of IgG3 mAbs, which have a lower affinity for the FcRn is approximately 7 days. Mabs which are Fc-deficient typically have an even shorter plasma half-life (e.g., 1.25 ± 0.63 h for blinatumomab in vivo), as they lack protection from degradation by the neonatal Fc receptor (FcRn) and in some cases also have a lower molecular weight than IgG, further increasing elimination through the kidneys [147,174][11][40]. It is conceivable that mAb internalization and FcRn-regulated release may affect the efficacy of a mAb if the dose of administration does not ensure that its free circulating fraction suffices to exert its action. Accordingly, blockade of the FcRn is therapeutically exploited to reduce the activity of pathogenic auto-antibodies (see rozanolixizumab, nipocalimab, batoclimab and efgartigimod in Section 6.5). A method to increase mAb half-life is to covalently attach a polyethylene glycol (PEG) chain to the mAb molecule (pegylation) as in the case of certolizumab pegol used for rheumatoid arthritis and Crohn’s disease [176][42].
The duration of biologic activity may differ substantially from their half-life because the former is primarily determined by the duration of the biological effects (e.g., the time required for a depleted cell population to recover). Consequently, the frequency of the administration depends on the mAb, its individual properties and the therapeutic strategy. Generally, mAbs are administered at fixed intervals, though in some cases dosing frequency may be determined by the duration of the effect as in the case of B cell depletion with rituximab treatment in multiple sclerosis (MS) and neuromyelitis optica spectrum disorders (NMOSD), where the peripheral blood CD19+ population may be used as a surrogate marker of B cells repopulation [177][43].
A notable drawback of using mAbs for neurological diseases is their low accessibility to the CNS compartment. The normal brain-to-blood IgG concentration ratio of IV infused mAbs is approximately 0.1%. The passage through the BBB could be facilitated by the use of bispecific Abs where one specificity recognizes a receptor at the BBB, which promotes transcytosis, and the other specificity recognizes a potential therapeutic target such as Aβ, tau or tumor-specific targets (Figure 3). The best studied receptors for targeting brain tissue and promoting passage through the BBB are the insulin receptor (InsR), the LDL-related protein type 1 (LRP1) and the transferrin receptor (TfR) [178,179][44][45]. Using bispecific Abs with BBB shuttle function has been shown to increase brain-to-blood IgG concentration ratio of IV infused mAbs to 2–3% [180][46]. Other methods to improve mAb delivery to the CNS compartment are also being explored [181][47].
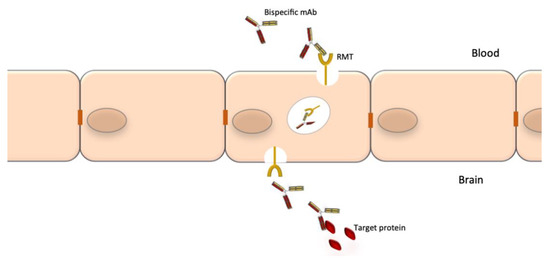
Figure 3. Crossing of the blood-brain barrier by specifically engineered bispecific mAbs. The blood-brain barrier (BBB) is an obstacle for the delivery of mAbs to the CNS. Bispecific mAbs may be used to cross the BBB as through their one specificity they can be shuttled via receptor-mediated transport (RMT), while the other can be used to bind the target proteins.
Interestingly, a recent double-blind trial investigated the effects of intrathecal and intravenous administration of rituximab versus placebo on a number of biomarkers of B cells depletion, inflammation and neurodegeneration in progressive MS (RIVITALISE trial; NCT01212094). The trial was discontinued early because at interim analysis, cerebrospinal fluid (CSF) B cells were only partially and transiently depleted and neurofilament light chain levels used as a marker of axonal damage were unchanged. The study identified low CSF levels of lytic complement factors and paucity of cytotoxic CD56dim NK cells as key contributors to decreased efficacy of intrathecally-administered rituximab [74][48].
5. Antibodies used in neurology
Antibodies of all types (murine, chimeric, humanized and human) have been approved by Food and Drug Administration (FDA), European Medicines Agency (EMA) and other national agencies for the treatment of several diseases. Since the approval of OKT3, the use of mAbs has progressively come to dominate therapeutics in all fields of medicine, including neurology. Many of the mAbs used in neurology today have been repurposed from their original indications for hematological neoplasias (e.g., alemtuzumab, ofatumumab and rituximab) or rheumatological disease (e.g., tocilizumab) [4,7,8,9][4][49][50][51]. Other mAbs have been developed originally for neurological disease (e.g., ocrelizumab for multiple sclerosis or mAbs for migraine prophylaxis). Sixteen marketed mAbs are used in neurology primarily for neuroimmunological conditions and migraine (Table 1). Nevertheless, many more mAbs are in development for neuroimmunological and neurodegenerative conditions (Table 2).
Table 1. Marketed monoclonal antibodies used in neurology.
Name | Type | Target | Action | Route | Neurological | Indication | Adverse Effects of Special Interest | References | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Alemtuzumab | humanized | IgG 1 | CD52 | Depletes CD52+ T and B cells | IV | RR-MS * | Infusion reactions | Secondary autoimmunity | Cerebrovascular accidents | |||||||||||||||||||||||
Bevacizumab | humanized | IgG 1 | VEGF | Inhibition of angiogenesis | IV | Glioblastoma * | hypertension, | gastrointestinal perforation, bleeding, PRES | ||||||||||||||||||||||||
Daclizumab | humanized | IgG 1 | IL2R-α (CD25) | Blocks the high affinity IL-2 receptor containing the α subunit | SC | RR-MS * | Autoimmune encephalitis, hepatitis and rashes |
[18,19,20,21,22,23,24,25,26,27] |
[60][61][62][63][64][65][66][67][68][69] |
|||||||||||||||||||||||
Eculizumab | humanized | IgG 2/4 | C5 complement protein | Inhibition of the terminal C5 complement pathway | IV | Anti-AChR Ab+ MG * | AQP-4 + NMOSD * | Meningococcal infections | ||||||||||||||||||||||||
Eptinezumab | humanized | IgG 1 | CGRP ligand | Selectively bind to isoforms a and b of CGRP | IV | EM* and CM * | Nasopharyngitis | Hypersensitivity reactions |
[34] |
[76] |
||||||||||||||||||||||
Erenumab | fully human | IgG 2 | CGRP receptor | Competitively and reversibly binds the CGRP receptor | SC | EM * and CM * | Constipation | Injection site reactions | ||||||||||||||||||||||||
Fremanezumab | humanized | IgG 2 | CGRP ligand | Selectively bind to isoforms a and b of CGRP | SC | EM * and CM * | Injection site reactions | |||||||||||||||||||||||||
Galcanezumab | humanized | IgG 4 | CGRP ligand | Binds CGRP and prevents its biological activity | SC | EM * and CM * | Cluster headache | Injection site reactions |
[42,43,44,[45,8646,47,][87][88][48] | |||||||||||||||||||||||
Inebilizumab | humanized | IgG 1 | CD19 | Depletes B cells and some short-lived plasmablasts and plasma cells | IV | AQP-4+ NMOSD | Infusion reactions, infections | |||||||||||||||||||||||||
Infliximab | chimeric | IgG 1 | TNF-α blockade | TNF-α signaling blockade | IV | DM/PM | Behcet disease | Neurosarcoidosis | Infusion reactions | CNS demyelination |
[29, | |||||||||||||||||||||
Natalizumab | humanized | IgG 4 | α4β1 integrin (CD49d) | Inhibits the entry of lymphocytes into the brain parenchyma | IV | RR-MS * | PML, | hepatotoxicity |
[63,20][64,9765,66,][98][99][100][101][102][103][104][105][106][107]67[108] |
|||||||||||||||||||||||
Ocrelizumab | humanized | IgG 1 | CD20 | Depletes B cells | IV | RR-MS * | PP-MS * | Infusion reactions, infections | ||||||||||||||||||||||||
Ofatumumab | fully human | IgG 1 | CD20 | Depletes B cells | SC | RR-MS * | Injections site reactions, infections, neutropenia | |||||||||||||||||||||||||
Rituximab | chimeric | IgG 1 | CD20 | Depletes B cells | IV | RR-MS | NMOSD; MG; CIDP; MMN, anti-MAG neuropathy | PM/DM | Infusion reactions | PML |
[4,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106] |
[4][26][48][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132][133][134][135][136][137][138][139][140][141][142][143][144][145] |
||||||||||||||||||||
Satralizumab | humanized | IgG 2 | IL-6 receptor | IL-6 receptor signaling blockade | SC | Anti-AQP4 Ab+ NMOSD * | Infections, neutropenia, elevated liver enzymes | |||||||||||||||||||||||||
Tocilizumab | humanized | IgG 1 | IL-6 receptor | IL-6 receptor signaling blockade | IV | NMOSD | CRS | Infusion reactions, | Infections |
*: officially approved indication, AQP4: aquaporin 4; CIDP: chronic inflammatory demyelinating polyneuropathy; CGRP calcitonin gene-related peptide; CNS: central nervous system; CM: chronic migraine; CRS: cytokine release syndrome; DM/PM: dermatomyositis/polymyositis; EM episodic migraine; IL-6R: interleukin 6 receptor; MG: myasthenia gravis; MMN: multifocal motor neuropathy; NMOSD: neuromyelitis optica spectrum disorder; PML: progressive multifocal leukoencephalopathy; PP-MS: primary progressive multiple sclerosis; PRES: posterior reversible encephalopathy syndrome; RR-MS: relapsing remitting multiple sclerosis; TNF-α: tumor necrosis factor-α.
Table 2. Monoclonal antibodies in development for various neurological indications.
Name | Type | Target | Action | Stage of Development | Neurological | Indication | References | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Aducanumab | (BIIB037) | fully human | IgG1 | Aβ | Binding of the aggregated Aβ forms | In phase IΙΙ | Prodromal to mild AD | ||||||||||||||||
Aquaporumab | fully human | (mutated Fc) | AQP-4 | Competitively inhibits binding of anti-AQP-4 auto-Abs | not yet in clinical trials | NMOSD | |||||||||||||||||
Batoclimab | (HBM9161) | fully human | IgG1 | FcRn | Reduction of auto-antibody levels | In phase II | MG |
[119] |
[158] |
||||||||||||||
Cinpanemab | (BIIB054) | humanized | IgG1 | α-synuclein | Prevention of accumulation and aggregation of α-synuclein | In phase II | PD | ||||||||||||||||
Donanemab | (N3pG) | humanized | IgG1 | Aβ | Binding aggregated Aβ forms | In phase II | Mild AD | ||||||||||||||||
Efgartigimod | Antibody fragment | FcRn | Reduction of auto-antibody levels | In phase II for CIDP | completed phase III for MG | MG | CIDP | ||||||||||||||||
Gantenerumab | (RG1450) | fully human | IgG1 | Aβ | Binding aggregated Aβ forms | In two phase III trials | Prodromal and mild AD | ||||||||||||||||
Gosuranemab | (BIIB092) | humanized | IgG4 | tau | Targeting abnormal forms of tau protein or soluble oligomers | In phase II | Prodromal to mild AD |
[169] |
|||||||||||||||
Nipocalimab | (M 281) | fully human | IgG1 | FcRn | Reduction of auto-antibody levels | Completed phase II trial | MG |
[132] |
|
||||||||||||||
Opicinumab | (BIIB033) | fully human IgG1 | LINGO-1 | Promotion of remyelination | In phase II | MS |
|
||||||||||||||||
Ravulizumab | (ALXN1210) | humanized IgG2/4 | C5 | Inhibition of the C5 terminal complement pathway | In phase III | AQP-4+ NMOSD, MG |
|
||||||||||||||||
Rilotumumab | (AMG102) | fully human IgG2 | HGF | Prevents activation of the c-Met receptor and tumor cell growth | In phase II | Glioblastoma |
[59] |
||||||||||||||||
Rozanolixizumab | (UCB 7665) | humanized IgG4 | FcRn | Reduction of auto-antibody levels | Completed a phase II study | MG |
[139] |
|
|||||||||||||||
Semorinemab | (RG6100) | humanized IgG4 | tau | Targeting all isoforms of tau protein | In phase II | Prodromal to mild AD |
[130] |
[169] |
|||||||||||||||
Tilavonemab | (ABBV 8E12) | humanized IgG4 | tau | Targeting abnormal extracellular forms of tau protein | In phase II | Prodromal to mild AD |
[130] |
[169] |
Aβ: amyloid beta peptide; AD: Alzheimer’s disease; AQP4: aquaporin 4; CIDP: chronic, FcRn: neonatal Fc receptor; HGF: hepatocyte growth factor; LINGO-1: Leucine rich repeat, Ig domain containing, Nogo receptor interactive protein-1; MG: myasthenia gravis; NMOSD: neuromyelitis optica spectrum disorder.
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497.
- Kung, P.; Goldstein, G.; Reinherz, E.L.; Schlossman, S.F. Monoclonal antibodies defining distinctive human T cell surface antigens. Science 1979, 206, 347–349.
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855.
- Grillo-López, A.J.; White, C.A.; Dallaire, B.K.; Varns, C.L.; Shen, C.D.; Wei, A.; Leonard, J.E.; McClure, A.; Weaver, R.; Cairelli, S.; et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000, 1, 1–9.
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525.
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1.
- Weiner, L.M.; Adams, G.P.; Von Mehren, M. Therapeutic monoclonal antibodies: General principles. In Cancer Principles and Practice and Oncology, 6th ed.; DeVita, V.T., Hellman, H., Rosenberg, S.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; pp. 495–508.
- Chirmule, N.; Jawa, V.; Meibohm, B. Immunogenicity to therapeutic proteins: Impact on PK/PD and efficacy. AAPS J. 2012, 14, 296–302.
- Van den Bemt, B.J.; Wolbink, G.J.; Hekster, Y.A.; van Riel, P.L.C.M.; Benraad, B.; van den Hoogen, F.H.J. Anti-infliximab antibodies are already detectable in most patients with rheumatoid arthritis halfway through an infusion cycle: An open-label pharmacokinetic cohort study. BMC Musculoskelet. Disord. 2011, 12, 12.
- Boulianne, G.L.; Hozumi, N.; Shulman, M.J. Production of functional chimaeric mouse/human antibody. Nature 1984, 312, 643–646.
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588.
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357.
- Steinitz, M. Three decades of human monoclonal antibodies: Past, present and future developments. Human. Antibodies 2009, 18, 1–10.
- Jespers, L.S.; Roberts, A.; Mahler, S.M.; Winter, G.; Hoogenboom, H.R. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology (NY) 1994, 12, 899–903.
- Strohl, W.R.; Strohl, L.M. 8-Monoclonal antibody targets and mechanisms of action. In Therapeutic Antibody Engineering; Series in Biomedicine; Woodhead Publishing Limited: Cambridge, UK, 2012; pp. 163–595. ISBN 9781907568374.
- Buss, N.A.; Henderson, S.J.; McFarlane, M.; Shenton, J.M.; de Haan, L. Monoclonal antibody therapeutics: History and future. Curr. Opin. Pharmacol. 2012, 12, 615–622.
- Zafir-Lavie, I.; Michaeli, Y.; Reiter, Y. Novel antibodies as anticancer agents. Oncogene 2007, 26, 3714–3733.
- Russell, F.A.; King, R.; Smillie, S.-J.; Kodji, X.; Brain, S.D. Calcitonin Gene-Related Peptide: Physiology and Pathophysiology. Physiol. Rev. 2014, 94, 1099–1142.
- Mitsikostas, D.D.; Rapoport, A.M. New players in the preventive treatment of migraine. BMC Med. 2015, 13, 279.
- Yu, Y.; Schürpf, T.; Springer, T.A. How natalizumab binds and antagonizes α4 integrins. J. Biol. Chem. 2013, 288, 32314–32325.
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011, 19, 101–113.
- Dobson, C.L.; Main, S.; Newton, P.; Chodorge, M.; Cadwallander, K.; Humphreys, R.; Albert, V.; Vaughan, T.J.; Minter, R.R.; Edwards, B.M. Human monomeric antibody fragments to TRAIL-R1 and TRAIL-R2 that display potent in vitro agonism. mAbs 2009, 1, 552–562.
- Suzuki, M.; Kato, C.; Kato, A. Therapeutic antibodies: Their mechanisms of action and the pathological findings they induce in toxicity studies. J. Toxicol. Pathol. 2015, 28, 133–139.
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520.
- Walport, M.J. Complement. N. Engl. J. Med. 2001, 344, 1058–1066.
- Golay, J.; Semenzato, G.; Rambaldi, A.; Foà, R.; Gaidano, G.; Gamba, E.; Pane, F.; Pinto, A.; Specchia, G.; Zaja, F.; et al. Lessons for the clinic from rituximab pharmacokinetics and pharmacodynamics. MAbs 2013, 5, 826–837.
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439.
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45.
- Teicher, B.A.; Chari, R.V. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397.
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. 90 Y-ibritumomab tiuxetan: A nearly forgotten opportunity. Oncotarget 2016, 7, 7597–7609.
- Turshudzhyan, A. The role of ado-trastuzumab emtansine in current clinical practice. J. Oncol. Pharm. Pract. 2021, 27, 150–155.
- Husain, B.; Ellerman, D. Expanding the Boundaries of Biotherapeutics with Bispecific Antibodies. BioDrugs 2018, 32, 441–464.
- Weber, F.; Bohrmann, B.; Niewoehner, J.; Fischer, J.A.A.; Rueger, P.; Tiefenthaler, G.; Moelleken, J.; Bujotzek, A.; Brady, L.; Singer, T.; et al. Brain shuttle antibody for Alzheimer’s disease with attenuated peripheral effector function due to an inverted binding mode. Cell Rep. 2018, 22, 149–162.
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug. Discov. 2019, 18, 585–608.
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. N. Engl. J. Med. 2017, 377, 809–818.
- He, P.; Xin, W.; Schulz, P.; Sierks, M.R. Bispecific Antibody Fragment Targeting APP and Inducing α-Site Cleavage Restores Neuronal Health in an Alzheimer’s Mouse Model. Mol. Neurobiol. 2019, 56, 7420–7432.
- Li, J.; Zhang, Z.; Lv, L.; Qiao, H.; Chen, X.; Zou, C. A bispecific antibody (ScBsAbAgn-2/TSPO) target for Ang-2 and TSPO resulted in therapeutic effects against glioblastomas. Biochem. Biophys. Res. Commun. 2016, 472, 384–391.
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/ VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481.
- Newsome, B.W.; Ernstoff, M.S. The clinical pharmacology of therapeutic monoclonal antibodies in the treatment of malignancy; have the magic bullets arrived? Br. J. Clin. Pharmacol. 2008, 66, 6.
- Loffler, A.; Kufer, P.; Lutterbüse, R.; Zettl, F.; Daniel, P.T.; Schwenkenbecher, J.M.; Riethmüller, G.; Dörken, B.; Bargou, R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 2000, 95, 2098–2103.
- Goebl, N.A.; Babbey, C.M.; Datta-Mannan, A.; Witcher, D.R.; Wroblewski, V.J.; Dunn, K.W. Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol. Biol. Cell. 2008, 19, 5490–5505.
- Goel, N.; Stephens, S. Certolizumab pegol. mAbs 2010, 2, 137–147.
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral CD19(+) B-cell counts and infusion intervals as a surrogate for long-term B-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67.
- Oller-Salvia, B.; Sanchez-Navarro, M.; Giralt, E.; Teixido, M. Blood-brain barrier shuttle peptides: An emerging paradigm for brain delivery. Chem. Soc. Rev. 2016, 45, 4690–4707.
- Sharma, G.; Lakkadwala, S.; Modgil, A.; Singh, J. The role of cell-penetrating peptide and transferrin on enhanced delivery of drug to brain. Int. J. Mol. Sci. 2016, 17, 806.
- Thom, G.; Hatcher, J.; Hearn, A.; Paterson, J.; Rodrigo, N.; Beljean, A.; Gurrell, I.; Webster, C. Isolation of blood-brain barrier-crossing antibodies from a phage display library by competitive elution and their ability to penetrate the central nervous system. MAbs 2018, 10, 304–314.
- Razpotnik, R.; Novak, N.; Curin, Šerbec, V.; Rajcevic, U. Targeting Malignant Brain Tumors with Antibodies. Front. Immunol. 2017, 8, 1181.
- Komori, M.; Lin, Y.C.; Cortese, I.; Blake, A.; Ohayon, J.; Cherup, J.; Maric, D.; Kosa, P.T.; Wu, T.; Bielekova, B. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 166–179.
- Warner, J.L.; Arnason, J.E. Alemtuzumab use in relapsed and refractory chronic lymphocytic leukemia: A history and discussion of future rational use. Ther. Adv. Hematol. 2012, 3, 375–389.
- Karlin, L.; Coiffier, B. Ofatumumab in the treatment of non-Hodgkin’s lymphomas. Expert Opin. Biol. Ther. 2015, 15, 1085–1091.
- Kaneko, A. Tocilizumab in rheumatoid arthritis: Efficacy, safety and its place in therapy. Ther. Adv. Chronic Dis. 2013, 4, 15–21.
- Coles, A.J.; Compston, D.A.S.; Selmaj, K.W.; Lake, S.L.; Moran, S.; Margolin, D.H.; Lake, S.L.; Moran, S.; Palmer, J.; Smith, M.S.; et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N. Engl. J. Med. 2008, 359, 1786–1801.
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828.
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomized controlled phase 3 trial. Lancet 2012, 380, 1829–1839.
- Food and Drug Administration: LEMTRADA (Alemtuzumab). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/103948s5158lbl.pdf (accessed on 20 November 2020).
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963.
- Chamberlain, M.C.; Johnston, S. Bevacizumab for recurrent alkylator-refractory anaplastic oligodendroglioma. Cancer 2009, 115, 1734–1743.
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: Efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787.
- Affronti, M.L.; Jackman, J.G.; McSherry, F.; Herndon, J.E., 2nd; Massey, E.C., Jr.; Lipp, E.; Desjardins, A.; Friedman, H.S.; Vlahovic, G.; Vredenburgh, J.; et al. Phase II Study to Evaluate the Efficacy and Safety of Rilotumumab and Bevacizumab in Subjects with Recurrent Malignant Glioma. Oncologist 2018, 23, 889-e98.
- Bielekova, B. Daclizumab Therapy for Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a034470.
- Gold, R.; Radue, E.W.; Giovannoni, G.; Selmaj, K.; Havrdova, E.K.; Montalban, X.; Stefoski, D.; Sprenger, T.; Robinson, R.R.; Fam, S.; et al. Long-term safety and efficacy of daclizumab beta in relapsing-remitting multiple sclerosis: 6-year results from the SELECTED open-label extension study. J. Neurol. 2020, 267, 2851–2864.
- Giovannoni, G.; Gold, R.; Selmaj, K.; Havrdova, E.; Montalban, X.; Radue, E.W.; Stefoski, D.; McNeill, M.; Amaravadi, L.; Sweetser, M.; et al. Daclizumab high-yield process in relapsing-remitting multiple sclerosis (SELECTION): A multicentre, randomised, double-blind extension trial. Lancet Neurol. 2014, 13, 472–481.
- Kappos, L.; Wiendl, H.; Selmaj, K.; Arnold, D.L.; Havrdova, E.; Boyko, A.; Kaufman, M.; Rose, J.; Greenberg, S.; Sweetser, M.; et al. Daclizumab HYP versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2015, 373, 1418–1428.
- Diao, L.; Hang, Y.; Othman, A.A.; Mehta, D.; Amaravadi, L.; Nestorov, I.; Tran, J.Q. Population PK-PD analyses of CD25 occupancy, CD56bright NK cell expansion, and regulatory T cell reduction by daclizumab HYP in subjects with multiple sclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1333–1342.
- European Medicines Agency. EMA Urgently Reviewing Multiple Sclerosis Medicine Zinbryta Following Cases of Inflammatory Brain Disorders; Press Release 02/03/2018. Available online: https://www.ema.europa.eu/en/news/ema-urgently-reviewing-multiple-sclerosis-medicine-zinbryta-following-cases-inflammatory-brain (accessed on 20 November 2020).
- Luessi, F.; Engel, S.; Spreer, A.; Bittner, S.; Zipp, F. GFAPalpha IgG-associated encephalitis upon daclizumab treatment of MS. Neurol. NeuroImmunol. Neuroinflamm. 2018, 5, e481.
- Avasarala, J. DRESS Syndrome and Daclizumab Failure-Were Potentially Dangerous Signs Missed in Clinical Trials? Drug Target. Insights 2018, 12, 1177392818785136.
- Cortese, I.; Ohayon, J.; Fenton, K.; Lee, C.C.; Raffeld, M.; Cowen, E.W.; DiGiovanna, J.J.; Bielekova, B. Cutaneous adverse events in multiple sclerosis patients treated with daclizumab. Neurology 2016, 86, 847–855.
- Cohan, S.L.; Lucassen, E.B.; Romba, M.C.; Linch, S.N. Daclizumab: Mechanisms of Action, Therapeutic Efficacy, Adverse Events and Its Uncovering the Potential Role of Innate Immune System Recruitment as a Treatment Strategy for Relapsing Multiple Sclerosis. Biomedicines 2019, 7, 18.
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625.
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264.
- Xue, T.; Yang, Y.; Lu, Q.; Gao, B.; Chen, Z.; Wang, Z. Efficacy and Safety of Monoclonal Antibody Therapy in Neuromyelitis Optica Spectrum Disorders: Evidence from Randomized Controlled Trials. Mult. Scler. Relat. Disord. 2020, 43, 102166.
- Akaishi, T.; Nakashima, I. Efficiency of antibody therapy in demyelinating diseases. Int. Immunol. 2017, 29, 327–335.
- Howard, J.F.; Utsugisawa, K.; Benatar, M.; Murai, H.; Barohn, R.J.; Illa, I.; Jacob, S.; Vissing, J.; Burns, T.M.; Kissel, J.T.; et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): A phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 2017, 16, 976–986.
- Nishimura, J.; Yamamoto, M.; Hayashi, S.; Ohyashiki, K.; Ando, K.; Brodsky, A.L.; Noji, H.; Kitamura, K.; Eto, T.; Takahashi, T.; et al. Genetic variants in C5 and poor response to eculizumab. N. Engl. J. Med. 2014, 370, 632–639.
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in episodic migraine: A randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020, 40, 241–254.
- Goadsby, P.J.; Reuter, U.; Hallström, Y.; Broessner, G.; Bonner, J.H.; Zhang, F.; Sapra, S.; Picard, H.; Mikol, D.D.; Lenz, R.A. A Controlled Trial of Erenumab for Episodic Migraine. N. Engl. J. Med. 2017, 377, 2123–2132.
- Ashina, M.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Dodick, D.W.; Xue, F.; Zhang, F.; Paiva da Silva Lima, G.; Cheng, S.; Mikol, D.D. Long-term efficacy and safety of erenumab in migraine prevention: Results from a 5-year, open-label treatment phase of a randomized clinical trial. Eur. J. Neurol. 2021.
- Dodick, D.W.; Ashina, M.; Brandes, J.L.; Kudrow, D.; Lanteri-Minet, M.; Osipova, V.; Palmer, K.; Picard, H.; Mikol, D.D.; Lenz, R.A. ARISE: A phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia 2018, 38, 1026–1037.
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo–controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434.
- Ashina, M.; Dodick, D.; Goadsby, P.J.; Reuter, U.; Silberstein, S.; Zhang, F.; Gage, J.R.; Cheng, S.; Mikol, D.D.; Lenz, R.A. Erenumab (AMG 334) in episodic migraine: Interim analysis of an ongoing open-label study. Neurology 2017, 89, 1237–1243.
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA 2018, 319, 1999–2008.
- Goadsby, P.J.; Silberstein, S.D.; Yeung, P.P.; Cohen, J.M.; Ning, X.; Yang, R.; Dodick, D.W. Long-term safety, tolerability, and efficacy of fremanezumab in migraine: A randomized study. Neurology 2020, 95, e2487–e2499.
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1080–1088.
- Skljarevski, V.; Matharu, M.; Millen, B.A.; Ossipov, M.H.; Kim, B.K.; Yang, J.Y. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia 2018, 38, 1442–1454.
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018, 91, e2211–e2221.
- Gklinos, P.; Mitsikostas, D.D. Galcanezumab in migraine prevention: A systematic review and meta-analysis of randomized controlled trials. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420918088.
- Goadsby, P.J.; Dodick, D.W.; Leone, M.; Bardos, J.N.; Oakes, T.M.; Millen, B.A.; Zhou, C.; Dowsett, S.A.; Aurora, S.K.; Ahn, A.H.; et al. Trial of galcanezumab in prevention of episodic cluster headache. N. Engl. J. Med. 2019, 381, 132–141.
- Camporeale, A.; Kudrow, D.; Sides, R.; Wang, S.; Van Dycke, A.; Selzler, K.J.; Stauffer, V.L. A phase 3, long-term, open-label safety study of Galcanezumab in patients with migraine. BMC Neurol. 2018, 18, 188.
- Martinez, J.M.; Hindiyeh, N.; Anglin, G.; Kalidas, K.; Hodsdon, M.E.; Kielbasa, W.; Moser, B.A.; Pearlman, E.M.; Garces, S. Assessment of immunogenicity from galcanezumab phase 3 trials in patients with episodic or chronic migraine. Cephalalgia 2020, 40, 978–989.
- Agius, M.A.; Klodowska-Duda, G.; Maciejowski, M.; Potemkowski, A.; Li, J.; Patra, K.; Wesley, J.; Madani, S.; Barron, G.; Katz, E.; et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult. Scler. 2019, 25, 235–245.
- Frampton, J.E. Inebilizumab: First Approval. Drugs 2020, 80, 1259–1264.
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234.
- Saadoun, S.; Waters, P.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010, 133, 349–361.
- Kuroda, R.; Suzuki, J.; Muramatsu, M.; Tasaki, A.; Yano, M.; Imai, N.; Serizawa, M.; Kobari, M. Efficacy of infliximab in neuro-Behçet’s disease presenting with isolated longitudinally extensive transverse myelitis. J. Neurol. 2013, 260, 3167–3170.
- Fritz, D.; Timmermans, W.M.C.; van Laar, J.A.M.; van Hagen, P.M.; Siepman, T.A.M.; van de Beek, D.; Brouwer, M.C. Infliximab treatment in pathology-confirmed neurosarcoidosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e847.
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910.
- Miller, D.H.; Soon, D.; Fernando, K.T.; MacManus, D.G.; Barker, G.J.; Yousry, T.A.; Fisher, E.; O’Connor, P.W.; Phillips, J.T.; Polman, C.H.; et al. AFFIRM Investigators. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology 2007, 68, 1390–1401.
- Butzkueven, H.; Kappos, L.; Wiendl, H.; Trojano, M.; Spelman, T.; Chang, I.; Kasliwal, R.; Jaitly, S.; Campbell, N.; Ho, P.R.; et al. Tysabri Observational Program (TOP) Investigators. Long-term safety and effectiveness of natalizumab treatment in clinical practice: 10 years of real-world data from the Tysabri Observational Program (TOP). J. Neurol. Neurosurg. Psychiatry 2020, 91, 660–668.
- Defer, G.; Mariotte, D.; Derache, N.; Toutirais, O.; Legros, H.; Cauquelin, B.; Le Mauff, B. CD49d expression as a promising biomarker to monitor natalizumab efficacy. J. Neurol. Sci. 2012, 314, 138–142.
- Vennegoor, A.; Rispens, T.; Strijbis, E.M.; Seewann, A.; Uitdehaag, B.M.; Balk, L.J.; Barkhof, F.; Polman, C.H.; Wolbink, G.; Killestein, J. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult. Scler. 2013, 19, 593–600.
- Jensen, P.E.; Koch-Henriksen, N.; Sellebjerg, F.; Sorensen, P.S. Prediction of antibody persistency from antibody titres to natalizumab. Mult. Scler. 2012, 18, 1493–1499.
- Lundkvist, M.; Engdahl, E.; Holmen, C.; Moverare, R.; Olsson, T.; Hillert, J.; Fogdell-Hahn, A. Characterization of anti-natalizumab antibodies in multiple sclerosis patients. Mult. Scler. 2013, 19, 757–764.
- Chisari, C.G.; Grimaldi, L.M.; Salemi, G.; Ragonese, P.; Iaffaldano, P.; Bonavita, S.; Sparaco, M.; Rovaris, M.; D’Arma, A.; Lugaresi, A.; et al. Clinical effectiveness of different natalizumab interval dosing schedules in a large Italian population of patients with multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1297–1303.
- Valenzuela, R.M.; Pula, J.H.; Garwacki, D.; Cotter, J.; Kattah, J.C. Cryptococcal meningitis in a multiple sclerosis patient taking natalizumab. J. Neurol. Sci. 2014, 340, 109–111.
- Dahdaleh, D.; Altmann, D.M.; Malik, O.; Nicholas, R.S. Breathlessness, night sweats, and weight loss on natalizumab. Lancet 2012, 380, 726–727.
- Rudick, R.A.; Stuart, W.H.; Calabresi, P.A.; Confavreux, C.; Galetta, S.L.; Radue, E.W.; Lublin, F.D.; Weinstock-Guttman, B.; Wynn, D.R.; Lynn, F.; et al. SENTINEL Investigators. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 911–923.
- Antezana, A.; Sigal, S.; Herbert, J.; Kister, I. Natalizumab-induced hepatic injury: A case report and review of literature. Mult. Scler. Relat. Disord. 2015, 4, 495–498.
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234.
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 2017, 376, 209–220.
- Sul, J.; Patel, A.; Gordon, M.L.; Steinklein, J.; Sanguinetti, S.; Pramanik, B.; Orban, Z.; Koralnik, I.; Harel, A. Progressive Multifocal Leukoencephalopathy in a Patient on Ocrelizumab Monotherapy. 62nd Annual meeting of the American Academy of Neurology (AAN), Toronto, Canada, Abstract S29.001. Neurology 2020, 94 (Suppl. 15), 4875.
- Ocrelizumab EMA Summary of Project Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/ocrevus-epar-product-information_en.pdf (accessed on 20 November 2020).
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557.
- Hauser, S.; Waubant, E.; Arnold, D.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688.
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471.
- Berntsson, S.G.; Kristoffersson, A.; Boström, I.; Feresiadou, A.; Burman, J.; Landtblom, A.M. Rapidly increasing off-label use of rituximab in multiple sclerosis in Sweden—Outlier or predecessor? Acta Neurol. Scand. 2018, 138, 327–331.
- Pellkofer, H.L.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Gerdes, L.A.; Havla, J.; Bittner, R.; Canis, M.; Meinl, E.; Hohlfeld, R.; et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011, 76, 1310–1315.
- Cree, B.A.C.; Lamb, S.; Morgan, K.; Chen, A.; Waubant, E.; Genain, C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005, 64, 1270–1272.
- Jacob, A.; Weinshenker, B.G.; Violich, I.; McLinskey, N.; Krupp, L.; Fox, R.J.; Wingerchuk, D.M.; Boggild, M.; Constantinescu, C.S.; Miller, A.; et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch Neurol. 2008, 65, 1443–1448.
- Kim, S.-H.; Kim, W.; Li, X.F.; Jung, I.-J.; Kim, H.J. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011, 68, 1412–1420.
- Bedi, G.S.; Brown, A.D.; Delgado, S.R.; Usmani, N.; Lam, B.L.; Sheremata, W.A. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult. Scler. 2011, 17, 1225–1230.
- Kim, S.H.; Huh, S.Y.; Lee, S.J.; Joung, A.; Kim, H.J. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013, 70, 1110–1117.
- Levine, T.D. Rituximab in the treatment of dermatomyositis. Arthritis Rheum. 2005, 52, 601–607.
- Kosmidis, M.; Dalakas, M. Practical considerations on the use of rituximab in autoimmune neurological disorders. Therap. Adv. Neurol. Disord. 2010, 3, 93–105.
- Oddis, C.V.; Reed, A.M.; Aggarwal, R.; Rider, L.G.; Ascherman, D.P.; Levesque, M.C.; Barohn, R.J.; Feldman, B.M.; Harris-Love, M.O.; Koontz, D.C.; et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum 2013, 65, 314e24.
- Aggarwal, R.; Bandos, A.; Reed, A.M.; Ascherman, D.P.; Barohn, R.J.; Feldman, B.M.; Miller, F.W.; Rider, L.G.; Harris-Love, M.O.; Levesque, M.C.; et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 2014, 66, 740e9.
- Aggarwal, R.; Loganathan, P.; Koontz, D.; Qi, Z.; Reed, A.M.; Oddis, C.V. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatol 2017, 56, 247e54.
- Brauner, S.; Eriksson-Dufva, A.; Albert Hietala, M.; Frisell, T.; Press, R.; Piehl, F. Comparison between rituximab treatment for new-onset generalized myasthenia gravis and refractory generalized myasthenia gravis. JAMA Neurol. 2020, e200851.
- Beecher, G.; Anderson, D.; Siddiqi, Z.A. Rituximab in refractory myasthenia gravis: Extended prospective study results. Muscle Nerve. 2018, 58, 452–455.
- Illa, I.; Diaz-Manera, J.; Rojas-Garcia, R.; Pradas, J.; Rey, A.; Blesa, R.; Juarez, C.; Gallardo, E. Sustained response to rituximab in anti-AchR and anti-MuSKpositive myasthenia gravis patients. J. Neuroimmun. 2008, 201–202, 90–94.
- Iorio, R.; Damato, V.; Alboini, P.E.; Evoli, A. Efficacy and safety of rituximab for myasthenia gravis: A systematic review and meta-analysis. J. Neurol. 2015, 262, 1115–1119.
- Tandan, R.; Hehir, M.K.; Waheed, W.; Howard, D.B. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve 2017, 56, 185–196.
- Hehir, M.K.; Hobson-Webb, L.D.; Benatar, M. Rituximab as treatment for anti-MuSK myasthenia gravis. Neurology 2017, 89, 1069–1077.
- Stieglbauer, K.; Pihler, R.; Topakian, R. 10-year-outcomes after rituximab for myasthenia gravis: Efficacy, safety, costs of in hospital care, and impact on childbearing potential. J. Neurol. Sci. 2017, 375, 241–244.
- Dos Santos, A.; Noury, J.-B.; Genetest, S.; Nadaj-Pakleza, A.; Cassereau, J.; Baron, C.; Videt, D.; Michel, L.; Pereon, Y.; Wiertlewki, S.; et al. Efficacy and safety of Rituximab in myasthenia gravis: A French multicentre real-life study. Eur. J. Neurol. 2020.
- Dıaz-Manera, J.; Martınez-Hernandez, E.; Querol, L.; Klooster, R.; Rojas-García, R.; Suárez-Calvet, X.; Muñoz-Blanco, J.L.; Mazia, C.; Straasheijm, K.R.; Gallardo, E.; et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012, 78, 189–193.
- Marino, M.; Basile, U.; Spagni, G. long-lasting rituximab-induced reduction of specific—But not total—IgG4 in MuSK-positive myasthenia gravis. Front. Immunol. 2020, 11, 613.
- Lebrun, C.; Bourg, V.; Bresch, S.; Cohen, M.; Rosenthal-Allieri, M.A.; Desnuelle, C. Ticchioni, Therapeutic target of memory B cells depletion helps to tailor administration frequency of rituximab in myasthenia gravis. J. Neuroimmunol. 2016, 298, 79–81.
- Ruegg, S.; Fuhr, P.; Steck, A. Rituximab stabilizes multifocal motor neuropathy increasingly less responsive to IVIg. Neurology 2004, 63, 2178–2179.
- Gorson, K.C.; Natarajan, N.; Ropper, A.H.; Weinstein, R. Rituximab treatment in patients with IVIg-dependent immune polyneuropathy: A prospective pilot trial. Muscle Nerve 2007, 35, 66–69.
- Dalakas, M.; Rakocevic, G.; Salajegheh, M.; Dambrosia, J.; Hahn, A.; Raju, R.; McElroy, B. Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein antibody demyelinating neuropathy. Ann. Neurol. 2009, 65, 286–293.
- Léger, J.M.; Viala, K.; Nicolas, G.; Créange, A.; Vallat, J.M.; Pouget, J.; Clavelou, P.; Vial, C.; Steck, A.; Musset, L.; et al. RIMAG Study Group (France and Switzerland). Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy. Neurology 2013, 80, 2217–2225.
- Gazzola, S.; Delmont, E.; Franques, J.; Boucraut, J.; Salort-Campana, E.; Verschueren, A.; Sagui, E.; Hubert, A.M.; Pouget, J.; Attarian, S. Predictive factors of efficacy of rituximab in patients with anti-MAG neuropathy. J. Neurol. Sci. 2017, 377, 144–148.
- Muley, S.A.; Jacobsen, B.; Parry, G.; Usman, U.; Ortega, E.; Walk, D.; Allen, J.; Pasnoor, M.; Varon, M.; Dimachkie, M.M. Rituximab in refractory chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2020, 61, 575–579.
- Van Vollenhoven, R.F.; Emery, P.; Bingham, C.O., 3rd; Keystone, E.C.; Fleischmann, R.M.; Furst, D.E.; Tyson, N.; Collinson, N.; Lehane, P.B. Long-term safety of rituximab in rheumatoid arthritis: 9.5-year follow-up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann. Rheum. Dis. 2013, 72, 1496–1502.
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: A randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet. Neurol. 2020, 19, 402–412.
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 2114–2124.
- Araki, M.; Aranami, T.; Matsuoka, T.; Nakamura, M.; Miyake, S.; Yamamura, T. Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod. Rheumatol. 2013, 23, 827–831.
- Kieseier, B.C.; Stüve, O.; Dehmel, T.; Goebels, N.; Leussink, V.I.; Mausberg, A.K.; Ringelstein, M.; Turowski, B.; Aktas, O.; Anoch, G.; et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: Implication for cellular immune responses. JAMA Neurol. 2012, 70, 390–393.
- ClinicalTrials.gov. Tocilizumab in the Treatment of Refractory Polymyositis and Dermatomyositis (TIM). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02043548,NCT02043548 (accessed on 20 November 2010).
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56, Update in: Nature 2017, 546, 564.
- Selkoe, D.J. Alzheimer disease and aducanumab: Adjusting our approach. Nat. Rev. Neurol. 2019, 15, 365–366.
- ClinicalTrials.gov. 2020.221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease (EMERGE). Available online: https://clinicaltrials.gov/ct2/show/NCT02484547,NCT02484547 (accessed on 20 November 2020).
- Howard, R.; Liu, K.Y. Questions EMERGE as Biogen claims aducanumab turnaround. Nat. Rev. Neurol. 2020, 16, 63–64.
- Schneider, L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol. 2020, 19, 111–112.
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012, 71, 314–322.
- Duan, T.; Tradtrantip, L.; Phuan, P.W.; Bennett, J.L.; Verkman, A.S. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 2020, 162, 107827.
- Clinical trials.gov. A Study to Evaluate the Efficacy, Safety and PD and PK of HBM9161 in MG Patients. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04346888?term=HBM9161&draw=2&rank=2 (accessed on 20 November 2020).
- Kwon, S.; Iba, M.; Kim, C.; Masliah, E. Immunotherapies for Aging-Related Neurodegenerative Diseases-Emerging Perspectives and New Targets. Neurotherapeutics 2020, 17, 935–954.
- Kuchimanchi, M.; Monine, M.; Kandadi Muralidharan, K.; Woodward, C.; Penner, N. Phase II Dose Selection for Alpha Synuclein-Targeting Antibody Cinpanemab (BIIB054) Based on Target Protein Binding Levels in the Brain CPT. Pharmacomet. Syst. Pharmacol. 2020.
- Logovinsky, V.; Satlin, A.; Lai, R.; Swanson, C.; Kaplow, J.; Osswald, G.; Basun, H.; Lannfelt, L. Safety and tolerability of BAN2401—A clinical study in Alzheimer’s disease with a protofibril selective Aβ antibody. Alzheimers Res. Ther. 2016, 8, 14.
- EISAI 2020 News Release. Initiation of New Phase III Clinical Study (ahead 3-45) of Ban2401 Preclinical (Asymptomatic) ALZHEIMER’S Disease. Available online: https://www.eisai.com/news/2020/news202042.html (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study to Evaluate Safety, Tolerability, and Efficacy of Lecanemab in Subjects with Early Alzheimer’s Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT01767311,NCT01767311 (accessed on 20 November 2020).
- Howard, J.F., Jr.; Bril, V.; Burns, T.M.; Mantegazza, R.; Bilinska, M.; Szczudlik, A.; Beydoun, S.; Garrido, F.J.R.R.; Piehl, F.; Rottoli, M.; et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 2019, 92, e2661–e2673.
- ClinicalTrials.gov. A Safety and Tolerability Study of ARGX-113 in Patients with Myasthenia Gravis Who Have Generalized Muscle Weakness. (ADAPT+). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03770403,NCT03770403 (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study to Assess the Safety and Efficacy of a Subcutaneous Formulation of Efgartigimod in Adults with Chronic Inflammatory Demyelinating Polyneuropathy (CIDP, an Autoimmune Disorder That Affects the Peripheral Nerves) (ADHERE). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04281472,NCT04281472 (accessed on 20 November 2020).
- ClinicalTrials.gov. A Study of Gantenerumab in Participants with Mild Alzheimer Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02051608?term=gantenerumab&draw=2&rank=9 (accessed on 20 November 2020).
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. SCarlet RoAD Investigators. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95.
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554.