Glioblastoma (GBM) is the most common form of primary malignant brain tumor with a devastatingly poor prognosis. The disease does not discriminate, affecting adults and children of both sexes, and has an average overall survival of 12–15 months, despite advances in diagnosis and rigorous treatment with chemotherapy, radiation therapy, and surgical resection. In addition, most survivors will eventually experience tumor recurrence that only imparts survival of a few months. GBM is highly heterogenous, invasive, vascularized, and almost always inaccessible for treatment. Based on all these outstanding obstacles, there have been tremendous efforts to develop alternative treatment options that allow for more efficient targeting of the tumor including small molecule drugs and immunotherapies. A number of other strategies in development include therapies based on nanoparticles, light, extracellular vesicles, and micro-RNA, and vessel co-option. Advances in these potential approaches shed a promising outlook on the future of GBM treatment.
- glioblastoma
- GBM pathogenesis
- heterogeneity
- targeted therapy
- immunotherapy
1. Introduction
Glioblastoma (GBM) is one of the most common forms of primary malignant brain tumor and has a very poor prognosis with an average patient survival lasting only 12–15 months [1][2]. This bleak outlook is due in part to the challenges that are presented by the anatomical location of the tumor as well as the heterogeneity of GBM cells and their rapid growth rate [3][4]. Although GBM is known to affect both adults and children, the incidence of GBM increases with age peaking in the 1970s [5]. Cancer incidence is roughly 2-3 individuals per 100,000 cases each year in the United States, with rates increasing slightly based on patient age [6][7][8]. There is also a slightly higher rate of incidence in men versus women, with men being 1.6 times more likely to develop GBM [9]. GBM accounts for approximately 46% of all diagnosed brain tumors and causes around 2.7% of all cancer-related deaths [3]. In fact, it is ranked as the third most common cause of death from cancer in patients between 15 and 34 years [7]. There are currently four grades of gliomas classified by the World Health Organization (grades I-IV) [7][10]. Grade IV gliomas are the most aggressive and invasive forms and are responsible for the poorest prognoses [10][11]. GBM typically refers to these grade IV gliomas and can be subdivided into primary and secondary types [12][13]. Although primary and secondary gliomas share similar histological characteristics, they have very different genetic profiles [14]. Primary GBM constitutes approximately 90% of GBM cases and is considered a de novo pathway of multistep tumorigenesis from glial cells while secondary GBM develops from lower-grade and pre-existing tumors such as diffuse astrocytomas [15]. Of the two, primary GBM is generally found to be more malignant than secondary GBM [16], and men are somewhat more likely to present with primary GBM while women are more likely to be diagnosed with secondary GBM [17].
The standard treatment options for GBM include surgery, chemotherapy, and radiation. However, even with these interventions, GBM still carries a dismal prognosis [18][19]. Diverse pathogenetic features and immunosuppression are two major contributors of current treatment failure. Although many studies have attempted to design effective treatments around these challenges, none have been developed that are capable of achieving long-term patient survival without causing unwanted damage to the delicate cells and neuronal tissues of the brain [4]. Over the past several years, targeted therapies and immunotherapies have shown great achievement in GBM management with promising results in clinical trials [18][19][20][21]. Other therapies in development include nanotechnology-based innovations, photodynamic strategies, gene therapy, and local destruction of the tumor via genetically modified bacteria or controlled hyperthermia. In this review, we discuss the current understanding of GBM’s pathogenetic features (i.e., cellular, molecular, and immunosuppressive properties) that contribute to treatment resistance. We also outline novel targeted therapies, different immunotherapeutic approaches, and a number of other promising/emerging treatment strategies for adult GBM that are currently under development.
2. Current Treatment
2.1. Standard of Care and Other FDA Approved Treatments
The current standards of care for GBM include maximal resection surgery, radiation, and temozolomide (TMZ) therapy—with TMZ and radiation being commenced within 30 days post-surgery [22][23][24]. Unfortunately, GBM response to TMZ varies between patients, and many types of GBM carry resistance to the compound. Treatment resistance to standard therapies is likely due to a combination of upregulated DNA repair mechanisms and the presence of GSCs that maintain an ability to self-renew and differentiate [25]. TMZ resistance also appears to be driven by the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT) that repairs DNA alkylation since patients bearing MGMT genes with methylated promoters seem to be more responsive to TMZ treatment [25]. However, TMZ damages both tumor and normal cells and does not eliminate GBM, so, options for alternative treatments are desperately needed [4].
Other treatments approved by the FDA for use in GBM therapy include bevacizumab and tumor-treating fields (TTFs). Bevacizumab is a humanized monoclonal antibody (mAb) that targets the angiogenic factor VEGF and was the first anti-angiogenic drug approved for patient use after showing increased overall survival in colorectal and non-small-cell lung cancers when combined with chemotherapy [26][27]. In addition, the antibody was observed to be safe in patients and mediate effective anti-tumor responses in Phase II clinical trial for recurrent GBM when combined with the chemotherapeutic drug irinotecan [28]. However, in two Phase III trials conducted in newly diagnosed GBM patients, treatment with bevacizumab in addition to either radiation or radiation plus TMZ showed no significant difference in overall survival compared to the placebo [29][30]. Although progression-free survival was better with treatment, patients suffered a higher frequency of adverse events and poorer quality of life [30].
TTFs are a non-invasive and anti-mitotic FDA-approved strategy for newly diagnosed cases of GBM (i.e., as adjuvant therapy) or recurring disease [31][32]. It involves using alternating electrical fields with a frequency range of 100-300 kHz and an intensity of 1 to 3 V/cm to interfere with the functions of rapidly dividing cancer cells, causing cessation of cell division and ultimately leading to cell death [31][33]. The theory behind this treatment is that the electrical fields create space between the growing ends of microtubules and tubulin dimers, thus, interfering with microtubule polymerization of the mitotic spindle [32]. Recently, TTF has been tested in combination with current standard-of-care in newly diagnosed GBM patients. Concurrent administration of TTF/radiation/TMZ followed by adjuvant TMZ/TTF demonstrated safety and promising preliminary efficacy [34], which warrants further clinical investigation in a larger patient cohort. In fact, a clinical study is currently ongoing utilizing this triple combination in 60 newly diagnosed GBM patients (NCT03869242).
2.2. Hurdles with Current Treatments
There are many aspects that make GBM difficult to effectively treat [35]. One complication is enabling the treatment drug to cross the BBB and reach the tumor [23]. It was previously thought that the BBB was uniformly disrupted in cases of GBM, and was, therefore, not an issue when designing treatment plans [23]. However, recent evidence suggests that a large portion of the BBB remains intact, presenting a challenge to many drug therapies [23]. Considering that drug molecules are unable to reach the tumor to potentiate effects, BBB transporters often remove most of the molecules that do manage to pass through [4].
The infiltrative and invasive growth of GBM also impedes complete surgical resection of tumor cells. Thus, secondary treatment is usually needed following surgery [36][37]. In addition, most GBMs that initially respond well to treatment recur after a period of a few months. Relapsed tumors generally have an even poorer overall survival and do not respond well to previously used treatments [38] as they acquire new mutations and evasive properties [39]. Tremendous efforts have been made to target those mutations by targeted therapies (discussed in more detail below).
Other major reasons for treatment failure can include: (i) GBM is extremely immunosuppressive [40][41][42], (ii) tumor cells contain a low somatic mutational load [43][44], which could explain poor responses to immune checkpoint blockade to the anti-PD-1 antibody (CheckMate-143), and (iii) presence of GSCs that help drive resistance to radiotherapy [45][46], chemotherapy [47], and anti-VEGF therapy [48]. Although TMZ is effective against MGMT-negative GSCs [49], the drug is incapable of eliminating MGMT-positive GSCs [50]. Resistance to TMZ in particular and other therapies mediated by GSCs also rely on an ability to regulate various miRNA molecules that can remodel different signaling pathways in response to treatment [51][52]. GSC plasticity also allows differentiation into a slow-cycling and persistent cellular state that can escape cytotoxicity from different targeted therapies [53]. Treatment-resistant GSCs further induce immunosuppression by recruiting M2-like tumor-associated macrophages (TAMs) and Tregs into the TME [54][55]. Lastly (iv), while effective anti-tumor immunity in GBM is profoundly inhibited, possibly by promoting subsets of dysfunctional T cells through various mechanisms [56], it is important to note that current standard therapies (including TMZ and high-dose corticosteroids) might worsen GBM’s immunosuppressive status [57][58][59]. Thus, there is a need to develop newer forms of immunotherapy that overcome immunosuppression and boost the host’s anti-tumor immune responses [36].
3. Treatments in Development
3.1. Targeted Therapies
Based on dysregulated signaling in GBM, targeted therapies are mainly categorized to ablate: the p53, RB, and receptor tyrosine kinase (RTK) signaling pathways (Figure 2). In general, intra- and inter-tumoral heterogeneity of mutated signaling pathways in GBM incite resistant mechanisms to monotherapeutic treatment with targeted agents. While combination therapies to target multiple pathways is one potential route to overcoming resistance, developing improved better strategies to impact each individual mutational alteration in GBM are gaining interest [36].
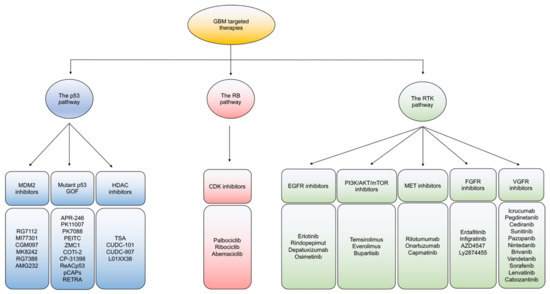
Figure 2. An overview of targeted therapies in GBM. Classification of current targeted therapies in GBM according to the three main signaling pathway alterations of the P53, Rb, and RTK pathways.
3.2. Immunotherapy
3.2.1. Immune Checkpoint Inhibitors (ICIs)
Anti-PD-1/PD-L1 Antibodies
A number of newer therapies in development constitute a major area in immunotherapy (Figure 3) [20][21][60][61][62][63]. GBM is an immunosuppressive tumor, with varying degrees of PD-L1 expression in tumor cells in GBM patients ranging from 61% to 88% [64]. Blocking PD-1/PD-L1 interactions unleash anti-tumor immune responses in various cancers such as melanoma. In GBM, although preclinical experience with PD-1/PD-L1 inhibition is quite promising [65][66][67][68], the clinical outcome of PD-1 blockade has been disappointing. For instance, the first large-scale randomized trial in recurrent GBM (CheckMate-143) revealed no significant difference in overall survival between patients receiving bevacizumab or nivolumab (an anti-PD-1 mAb), leading to premature termination of the nivolumab arm [69]. The inability of ICIs to cross the BBB, reduced frequency of immune infiltrates in the GBM TME, and a high level of GBM immunosuppression are considered major contributors of treatment failure for this approach in general [69]. In addition, the TME of PD-1 blockade non-responders is enriched with PTEN mutations regardless of GBM subtype, suggesting that combined targeting of PTEN and PD-1 could provide additive treatment benefits for this lethal disease [70]. Although adjuvant monotherapy of anti-PD-1 mAb failed to generate effective anti-tumor immunity, neoadjuvant PD-1 blockade led to the activation of GBM-specific T cells and downregulation of genes associated with the tumor cell-cycle. Therefore, timing of anti-PD-1/PD-L1 interventions in patients is probably crucial for mediating objective response rates and managing GBM [71].
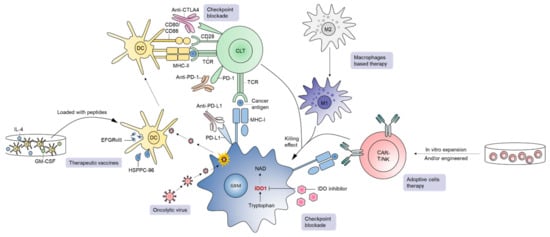
Figure 3. A brief overview of immunotherapies in GBM. Current immunotherapeutic approaches in GBM include checkpoint blockade, oncolytic virus, therapeutic vaccines, adoptive cell therapy, and macrophage-based strategies.
3.2.2. Oncolytic Viruses (OVs)
Oncolytic viruses (OVs) are a major developmental therapy of interest and have gained success in different cancer types, including GBM [55][61][72][73][74][75][76][77][78][79][80][81][82][83][84]. The recent FDA approval of an oncolytic herpes simplex virus (oHSV) (designated talimogene laherparepvec) has further fueled the field of oncolytic virotherapy. Tumor cells are noticeably distinct from normal cells by adopting behaviors such as increased proliferation and vascularization. OVs selectively infect tumor cells, killing them following replication, while leaving normal cells unscathed. At the same time, OVs trigger a cascade of anti-tumor immune responses [85], including increased tumor infiltration of immune cells [79][86]. OVs including oHSVs have shown promising efficacy in preclinical models of GBM [79][87][88] as well as in GBM patients [89]. In a recently published case study, four previously treated GBM patients received individualized treatment regimens comprised of three OVs (wild-type Newcastle disease virus [NDV], wild-type parvovirus [PV], and wild-type vaccinia virus [VV]). OVs were sequentially administered using the same catheter with a dose of 109 TCID50 for each virus in a volume of 10 mL and demonstrated impressive clinical and radiological responses with long-term survival up to 14 years [90]. OV-induced anti-tumor immunity can be further enhanced through OV-mediated expression of various cytokines/chemokines and immunomodulatory molecules [91]. OVs can also be used in combination with standard of care TMZ that produces synergistic anti-tumor effects in various preclinical cancer models including GBM [92][93][94][95][96][97]. However, a recent preclinical combination study (OV+TMZ) in GBM demonstrated conflicting results. Concurrent OV and TMZ therapy antagonized the anti-tumor properties of oncolytic virotherapy [80], indicating that co-applied administration of OV and TMZ represent a failed synergistic strategy as opposed to the pre-clinical benefits observed when TMZ was administered either before or after OV treatment [95][97][98]. Altogether, OVs serve to beneficially alter the TME to increase tumor immunogenicity, and synergize with ICIs [61][99]. Newer clinical studies are aiming to combine OVs and ICIs in order to improve patient outcomes as listed in Table 3 [99].
3.2.3. Therapeutic Vaccines
Vaccines are an active form of immunotherapy that has recently gained interest for GBM treatment [100][101]. The antigens such as EGFRvIII, heat shock protein (HSP), and any tumor-derived antigens can be loaded to DCs to incite immune responses against GBM [101]. In a randomized Phase II clinical trial in patients with relapsed EGFRvIII+ GBM, the EGFRvIII vaccine (designated Rindopepimut or CDX-110) delivered intradermally with GM-CSF (NCT01498328) resulted in the induction of EGFRvIII-specific immune responses, encouraging PFS and OS, and a significant extension of survival when the vaccine was administered in combination with bevacizumab [102]. The promising results of this trial led to a Phase III trial with Rindopepimut/GM-CSF in patients with newly diagnosed GBM, where all patients received standard-of-care TMZ (NCT01480479). Unfortunately, this Phase III trial was discontinued in early 2016 since Rindopepimut failed to significantly improve survival [103] and emphasizes the importance of identifying alternate and newer vaccine-based strategies to tackle GBM [104].
In contrast to EGFRvIII immunization that elicits immune responses to pre-defined tumor target, HSP vaccines offer immunity against a broad range of antigens. Induction of anti-tumor immunity against various antigenic targets is important to help minimize the outgrowth of target null variants, especially for cancer types that have high intra-tumoral heterogeneity like GBM [105]. The most well-known HSP vaccine is heat-shock protein peptide complex-96 (HSPPC-96) [106]. The safety and immunogenicity of HSPPC-96 monotherapy were demonstrated in a Phase I clinical trial in newly diagnosed GBM Patients [106]. HSPPC-96 is currently being tested in two separate Phase II clinical trials; one in combination with TMZ in patients with newly diagnosed GBM (NCT00905060) and the other in combination with bevacizumab in surgically resectable recurrent GBMs (NCT01814813) (Table 3).
DCs play a central role in linking innate and adaptive anti-tumor immune responses [107]. The principle of dendritic cell vaccines (DCV) is based on the ability of primed DCs to process/present tumor antigens and activate cytotoxic lymphocytes [108]. DCVs are prepared by isolating CD14+ monocytes from patient peripheral blood and further culturing cells ex vivo with granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin 4 (IL-4), and tumor antigens, prior to injecting the cells back into patients [109]. Although interest in DCV is further compelled by an FDA-approved DCV for the treatment of prostate cancer (Sipuleucel-T), most DCV-based clinical trials in GBM are still under phase I and II evaluations. For example, DCVax, an approved DCV for treatment of GBM in Switzerland, is currently being assessed in the US in patients with newly diagnosed GBM (NCT00045968) [110]. In a recent phase III study, DCVax was used alongside standard options and resulted in the extended survival of patients by 8 months compared to the control cohort [110][111]. Personalized neoantigen vaccine has also recently been tested in GBM clinical trials. For instance, in a Phase I/Ib study in newly diagnosed MGMT-unmethylated GBM, patients who did not receive dexamethasone had better neoantigen-specific CD4+ and CD8+ T cell responses with a higher number of TILs [112].
3.3. Nanomedicine
The neoplastic vasculature network is typically defective and leaky, which enhances the permeability and retention of nanoparticles in the TME [113][114]. Nanomedicine has been validated to enhance the efficacy of chemotherapies [115] and radiotherapy [116], but the safety and delivery of this approach have always been a major concern since nanoparticles preferentially deposit in the reticuloendothelial tissues of the kidneys, liver, and spleen [117][118]. Despite these issues, various anti-cancer nanoparticle therapies can produce superior efficacy versus non-nanoparticle formulation. For example, paclitaxel (PTX) or doxorubicin when administered as nanoparticles potentiate improved cytotoxicity against GBM compared to their parental compounds [119]. Several clinical trials are evaluating the therapeutic properties of different nanomedicine formulations such as nanoliposome (NCT00734682, NCT00944801, NCT01906385), Spherical Nucleic Acid (SNA) gold nanoparticles (NCT03020017), and nanocells (NCT02766699). To further advance this field in GBM and improve safety, an improved understanding of the long-term stability, biodistribution, and clearance mechanisms of nanoparticles is required.
References
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071.
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617.
- Lyon, J.G.; Mokarram, N.; Saxena, T.; Carroll, S.L.; Bellamkonda, R.V. Engineering challenges for brain tumor immunotherapy. Adv. Drug Deliv. Rev. 2017, 114, 19–32.
- Shergalis, A.; Bankhead, A., 3rd; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445.
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100.
- Lieberman, F. Glioblastoma update: Molecular biology, diagnosis, treatment, response assessment, and translational clinical trials. F1000Research 2017, 6, 1892.
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9.
- Melin, B.S.; Barnholtz-Sloan, J.S.; Wrensch, M.R.; Johansen, C.; Il’yasova, D.; Kinnersley, B.; Ostrom, Q.T.; Labreche, K.; Chen, Y.; Armstrong, G.; et al. Genome-wide association study of glioma subtypes identifies specific differences in genetic susceptibility to glioblastoma and non-glioblastoma tumors. Nat. Genet. 2017, 49, 789–794.
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci. Transl. Med. 2019, 11, eaao5253.
- Vigneswaran, K.; Neill, S.; Hadjipanayis, C.G. Beyond the World Health Organization grading of infiltrating gliomas: Advances in the molecular genetics of glioma classification. Ann. Transl. Med. 2015, 3, 95.
- Delgado-Martin, B.; Medina, M.A. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv. Sci (Weinh) 2020, 7, 1902971.
- Li, R.; Li, H.; Yan, W.; Yang, P.; Bao, Z.; Zhang, C.; Jiang, T.; You, Y. Genetic and clinical characteristics of primary and secondary glioblastoma is associated with differential molecular subtype distribution. Oncotarget 2015, 6, 7318–7324.
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772.
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453.
- Lee, D.H.; Ryu, H.W.; Won, H.R.; Kwon, S.H. Advances in epigenetic glioblastoma therapy. Oncotarget 2017, 8, 18577–18589.
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8.
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996.
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419.
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472.
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in Immunotherapy for Glioblastoma Multiforme. J. Immunol. Res. 2017, 2017, 3597613.
- Tivnan, A.; Heilinger, T.; Lavelle, E.C.; Prehn, J.H. Advances in immunotherapy for the treatment of glioblastoma. J. Neurooncol. 2017, 131, 1–9.
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12.
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191.
- Paolillo, M.; Boselli, C.; Schinelli, S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 2018, 8, 15.
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. (Tokyo) 2018, 58, 405–421.
- Rosen, L.S.; Jacobs, I.A.; Burkes, R.L. Bevacizumab in Colorectal Cancer: Current Role in Treatment and the Potential of Biosimilars. Target. Oncol. 2017, 12, 599–610.
- Russo, A.E.; Priolo, D.; Antonelli, G.; Libra, M.; McCubrey, J.A.; Ferrau, F. Bevacizumab in the treatment of NSCLC: Patient selection and perspectives. Lung Cancer (Auckl.) 2017, 8, 259–269.
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722.
- Kinzel, A.; Ambrogi, M.; Varshaver, M.; Kirson, E.D. Tumor Treating Fields for Glioblastoma Treatment: Patient Satisfaction and Compliance With the Second-Generation Optune((R)) System. Clin. Med. Insights Oncol. 2019, 13.
- Fabian, D.; Guillermo Prieto Eibl, M.D.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174.
- Burri, S.H.; Gondi, V.; Brown, P.D.; Mehta, M.P. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma: Guide for Oncologists. Am. J. Clin. Oncol. 2018, 41, 191–196.
- Bokstein, F.; Blumenthal, D.; Limon, D.; Harosh, C.B.; Ram, Z.; Grossman, R. Concurrent Tumor Treating Fields (TTFields) and Radiation Therapy for Newly Diagnosed Glioblastoma: A Prospective Safety and Feasibility Study. Front. Oncol. 2020, 10, 411.
- Noch, E.K.; Ramakrishna, R.; Magge, R. Challenges in the Treatment of Glioblastoma: Multisystem Mechanisms of Therapeutic Resistance. World Neurosurg. 2018, 116, 505–517.
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426.
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309.
- Chang, P.D.; Chow, D.S.; Yang, P.H.; Filippi, C.G.; Lignelli, A. Predicting Glioblastoma Recurrence by Early Changes in the Apparent Diffusion Coefficient Value and Signal Intensity on FLAIR Images. AJR Am. J. Roentgenol. 2017, 208, 57–65.
- Muscat, A.M.; Wong, N.C.; Drummond, K.J.; Algar, E.M.; Khasraw, M.; Verhaak, R.; Field, K.; Rosenthal, M.A.; Ashley, D.M. The evolutionary pattern of mutations in glioblastoma reveals therapy-mediated selection. Oncotarget 2018, 9, 7844–7858.
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11.
- Perng, P.; Lim, M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front. Oncol. 2015, 5, 153.
- Preusser, M.; Lim, M.; Hafler, D.A.; Reardon, D.A.; Sampson, J.H. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 2015, 11, 504–514.
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Goffart, N.; Lombard, A.; Lallemand, F.; Kroonen, J.; Nassen, J.; Di Valentin, E.; Berendsen, S.; Dedobbeleer, M.; Willems, E.; Robe, P.; et al. CXCL12 mediates glioblastoma resistance to radiotherapy in the subventricular zone. Neuro-Oncology 2017, 19, 66–77.
- Ventero, M.P.; Fuentes-Baile, M.; Quereda, C.; Perez-Valeciano, E.; Alenda, C.; Garcia-Morales, P.; Esposito, D.; Dorado, P.; Manuel Barbera, V.; Saceda, M. Radiotherapy resistance acquisition in Glioblastoma. Role of SOCS1 and SOCS3. PLoS ONE 2019, 14, e0212581.
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526.
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392.
- Beier, D.; Rohrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715.
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172.
- Tezcan, G.; Tunca, B.; Bekar, A.; Preusser, M.; Berghoff, A.S.; Egeli, U.; Cecener, G.; Ricken, G.; Budak, F.; Taskapilioglu, M.O.; et al. microRNA expression pattern modulates temozolomide response in GBM tumors with cancer stem cells. Cell Mol. Neurobiol. 2014, 34, 679–692.
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841.
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.
- Codony-Servat, J.; Rosell, R. Cancer stem cells and immunoresistance: Clinical implications and solutions. Transl. Lung Cancer Res. 2015, 4, 689–703.
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Curing glioblastoma: Oncolytic HSV-IL12 and checkpoint blockade. Oncoscience 2017, 4, 67–69.
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802.
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Cote, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro-Oncology 2011, 13, 393–400.
- Wang, S.; Yao, F.; Lu, X.; Li, Q.; Su, Z.; Lee, J.H.; Wang, C.; Du, L. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am. J. Cancer Res. 2019, 9, 1161–1171.
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front. Mol. Neurosci. 2019, 12, 65.
- Fisusi, F.A.; Schatzlein, A.G.; Uchegbu, I.F. Nanomedicines in the treatment of brain tumors. Nanomedicine (Lond.) 2018, 13, 579–583.
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786.
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D., Jr.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540.
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the immunotherapy of glioblastoma. Hum. Vaccin. Immunother. 2018, 14, 255–268.
- Caccese, M.; Indraccolo, S.; Zagonel, V.; Lombardi, G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit. Rev. Oncol. Hematol. 2019, 135, 128–134.
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349.
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136.
- Huang, B.Y.; Zhan, Y.P.; Zong, W.J.; Yu, C.J.; Li, J.F.; Qu, Y.M.; Han, S. The PD-1/B7-H1 pathway modulates the natural killer cells versus mouse glioma stem cells. PLoS ONE 2015, 10, e0134715.
- Jahan, N.; Talat, H.; Alonso, A.; Saha, D.; Curry, W.T. Triple combination immunotherapy with GVAX, anti-PD-1 monoclonal antibody, and agonist anti-OX40 monoclonal antibody is highly effective against murine intracranial glioma. Oncoimmunology 2019, 8, e1577108.
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794.
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486.
- Eissa, I.R.; Bustos-Villalobos, I.; Ichinose, T.; Matsumura, S.; Naoe, Y.; Miyajima, N.; Morimoto, D.; Mukoyama, N.; Zhiwen, W.; Tanaka, M.; et al. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers 2018, 10, 356.
- O’bryan, S.M.; Mathis, J.M. Oncolytic Virotherapy for Breast Cancer Treatment. Curr. Gene Ther. 2018, 18, 192–205.
- Ghouse, S.M.; Nguyen, H.M.; Bommareddy, P.K.; Guz-Montgomery, K.; Saha, D. Oncolytic Herpes Simplex Virus Encoding IL12 Controls Triple-Negative Breast Cancer Growth and Metastasis. Front. Oncol. 2020, 10, 384.
- Bommareddy, P.K.; Peters, C.; Saha, D.; Rabkin, S.D.; Kaufman, H.L. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Annu. Rev. Cancer Biol. 2018, 2, 155–173.
- Peters, C.; Paget, M.; Tshilenge, K.T.; Saha, D.; Antoszczyk, S.; Baars, A.; Frost, T.; Martuza, R.L.; Wakimoto, H.; Rabkin, S.D. Restriction of Replication of Oncolytic Herpes Simplex Virus with a Deletion of gamma34.5 in Glioblastoma Stem-Like Cells. J. Virol. 2018, 92.
- Saha, D.; Ahmed, S.S.; Rabkin, S.D. Exploring the Antitumor Effect of Virus in Malignant Glioma. Drugs Future 2015, 40, 739–749.
- Saha, D.; Martuza, R.L.; Curry, W.T. Viral oncolysis of glioblastoma. In Neurotropic Viral Infections, 2nd ed.; Reiss, C.S., Ed.; Springer: New York, NY, USA, 2016; Volume 2, pp. 481–517.
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.
- Saha, D.; Rabkin, S.D.; Martuza, R.L. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J. Immunother. Cancer 2020, 8, e000345.
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422.
- Siurala, M.; Havunen, R.; Saha, D.; Lumen, D.; Airaksinen, A.J.; Tahtinen, S.; Cervera-Carrascon, V.; Bramante, S.; Parviainen, S.; Vaha-Koskela, M.; et al. Adenoviral Delivery of Tumor Necrosis Factor-alpha and Interleukin-2 Enables Successful Adoptive Cell Therapy of Immunosuppressive Melanoma. Mol. Ther. 2016, 24, 1435–1443.
- Tahtinen, S.; Blattner, C.; Vaha-Koskela, M.; Saha, D.; Siurala, M.; Parviainen, S.; Utikal, J.; Kanerva, A.; Umansky, V.; Hemminki, A. T-Cell Therapy Enabling Adenoviruses Coding for IL2 and TNFalpha Induce Systemic Immunomodulation in Mice With Spontaneous Melanoma. J. Immunother. 2016, 39, 343–354.
- Vaha-Koskela, M.; Tahtinen, S.; Gronberg-Vaha-Koskela, S.; Taipale, K.; Saha, D.; Merisalo-Soikkeli, M.; Ahonen, M.; Rouvinen-Lagerstrom, N.; Hirvinen, M.; Veckman, V.; et al. Overcoming tumor resistance by heterologous adeno-poxvirus combination therapy. Mol. Ther. Oncol. 2015, 1, 14006.
- Saha, D.; Wakimoto, H.; Rabkin, S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016, 21, 26–34.
- Saha, D.; Rabkin, S.D. Immunohistochemistry for Tumor-Infiltrating Immune Cells After Oncolytic Virotherapy. Methods Mol. Biol. 2020, 2058, 179–190.
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479.
- Alayo, Q.A.; Ito, H.; Passaro, C.; Zdioruk, M.; Mahmoud, A.B.; Grauwet, K.; Zhang, X.; Lawler, S.E.; Reardon, D.A.; Goins, W.F.; et al. Glioblastoma infiltration of both tumor- and virus-antigen specific cytotoxic T cells correlates with experimental virotherapy responses. Sci. Rep. 2020, 10, 5095.
- Todo, T. ATIM-14. RESULTS OF PHASE II CLINICAL TRIAL OF ONCOLYTIC HERPES VIRUS G47Δ IN PATIENTS WITH GLIOBLASTOMA. Neuro-Oncology 2019, 21, vi4.
- Gesundheit, B.; Ben-David, E.; Posen, Y.; Ellis, R.; Wollmann, G.; Schneider, E.M.; Aigner, K.; Brauns, L.; Nesselhut, T.; Ackva, I.; et al. Effective Treatment of Glioblastoma Multiforme With Oncolytic Virotherapy: A Case-Series. Front. Oncol. 2020, 10, 702.
- Nguyen, H.M.; Guz-Montgomery, K.; Saha, D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells 2020, 9, 400.
- Alonso, M.M.; Gomez-Manzano, C.; Jiang, H.; Bekele, N.B.; Piao, Y.; Yung, W.K.; Alemany, R.; Fueyo, J. Combination of the oncolytic adenovirus ICOVIR-5 with chemotherapy provides enhanced anti-glioma effect in vivo. Cancer Gene Ther. 2007, 14, 756–761.
- Gomez-Gutierrez, J.G.; Nitz, J.; Sharma, R.; Wechman, S.L.; Riedinger, E.; Martinez-Jaramillo, E.; Sam Zhou, H.; McMasters, K.M. Combined therapy of oncolytic adenovirus and temozolomide enhances lung cancer virotherapy in vitro and in vivo. Virology 2016, 487, 249–259.
- Kanai, R.; Rabkin, S.D.; Yip, S.; Sgubin, D.; Zaupa, C.M.; Hirose, Y.; Louis, D.N.; Wakimoto, H.; Martuza, R.L. Oncolytic virus-mediated manipulation of DNA damage responses: Synergy with chemotherapy in killing glioblastoma stem cells. J. Natl. Cancer Inst. 2012, 104, 42–55.
- Kleijn, A.; van den Bossche, W.; Haefner, E.S.; Belcaid, Z.; Burghoorn-Maas, C.; Kloezeman, J.J.; Pas, S.D.; Leenstra, S.; Debets, R.; de Vrij, J.; et al. The Sequence of Delta24-RGD and TMZ Administration in Malignant Glioma Affects the Role of CD8(+)T Cell Anti-tumor Activity. Mol. Ther. Oncol. 2017, 5, 11–19.
- Garza-Morales, R.; Gonzalez-Ramos, R.; Chiba, A.; Montes de Oca-Luna, R.; McNally, L.R.; McMasters, K.M.; Gomez-Gutierrez, J.G. Temozolomide Enhances Triple-Negative Breast Cancer Virotherapy In Vitro. Cancers 2018, 10, 144.
- Pisklakova, A.; McKenzie, B.; Zemp, F.; Lun, X.; Kenchappa, R.S.; Etame, A.B.; Rahman, M.M.; Reilly, K.; Pilon-Thomas, S.; McFadden, G.; et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro-Oncology 2016, 18, 1088–1098.
- Bai, Y.; Chen, Y.; Hong, X.; Liu, X.; Su, X.; Li, S.; Dong, X.; Zhao, G.; Li, Y. Newcastle disease virus enhances the growth-inhibiting and proapoptotic effects of temozolomide on glioblastoma cells in vitro and in vivo. Sci. Rep. 2018, 8, 11470.
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncol. 2019, 13, 93–106.
- Dejaegher, J.; Van Gool, S.; De Vleeschouwer, S. Dendritic cell vaccination for glioblastoma multiforme: Review with focus on predictive factors for treatment response. Immunotargets Ther. 2014, 3, 55–66.
- Oh, T.; Sayegh, E.T.; Fakurnejad, S.; Oyon, D.; Lamano, J.B.; DiDomenico, J.D.; Bloch, O.; Parsa, A.T. Vaccine therapies in malignant glioma. Curr. Neurol. Neurosci. Rep. 2015, 15, 508.
- Reardon, D.A.; Schuster, J.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; Desjardins, A.; Ashby, L.S.; et al. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J. Clin. Oncol. 2015, 33, 2009.
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385.
- Binder, D.C.; Ladomersky, E.; Lenzen, A.; Zhai, L.; Lauing, K.L.; Otto-Meyer, S.D.; Lukas, R.V.; Wainwright, D.A. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Transl. Cancer Res. 2018, 7, S510–S513.
- Khansur, E.M.; Shah, A.H.; Lacy, K.; Kuchakulla, M.; Komotar, R.J. Novel Immunotherapeutics for the Treatment of Glioblastoma: The Last Decade of Research. Cureus 2018, 10, e2130.
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3, e99145.
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277.
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408.
- Nava, S.; Dossena, M.; Pogliani, S.; Pellegatta, S.; Antozzi, C.; Baggi, F.; Gellera, C.; Pollo, B.; Parati, E.A.; Finocchiaro, G.; et al. An optimized method for manufacturing a clinical scale dendritic cell-based vaccine for the treatment of glioblastoma. PLoS ONE 2012, 7, e52301.
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142.
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751.
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239.
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392.
- Karathanasis, E.; Ghaghada, K.B. Crossing the barrier: Treatment of brain tumors using nanochain particles. WIREs Nanomed. Nanobiotechnol. 2016, 8, 678–695.
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991.
- Hainfeld, J.F.; Ridwan, S.M.; Stanishevskiy, Y.; Panchal, R.; Slatkin, D.N.; Smilowitz, H.M. Iodine nanoparticles enhance radiotherapy of intracerebral human glioma in mice and increase efficacy of chemotherapy. Sci. Rep. 2019, 9, 4505.
- Samec, N.; Zottel, A.; Videtic Paska, A.; Jovcevska, I. Nanomedicine and Immunotherapy: A Step Further towards Precision Medicine for Glioblastoma. Molecules 2020, 25, 490.
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Preat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372.
- Walter, K.A.; Cahan, M.A.; Gur, A.; Tyler, B.; Hilton, J.; Colvin, O.M.; Burger, P.C.; Domb, A.; Brem, H. Interstitial taxol delivered from a biodegradable polymer implant against experimental malignant glioma. Cancer Res. 1994, 54, 2207–2212.