Pancreatic Ductal Adenocarcinoma (PDAC) is a fatal disease with poor prognosis because pa-tients rarely express symptoms in initial stages, which prevents early detection and diagnosis. Syndecans, a subfamily of proteoglycans, are involved in many physiological processes includ-ing cell proliferation, adhesion, and migration. Syndecans are physiologically found in many cell types and their interactions with other macromolecules enhance many pathways. In particu-lar, extracellular matrix components, growth factors, and integrins collect the majority of syndecans associations acting as biochemical, physical, and mechanical transducers. Syndecans are transmembrane glycoproteins, but occasionally their extracellular domain can be released from the cell surface by the action of matrix metalloproteinases, converting them into soluble molecules that are capable of binding distant molecules such as extracellular matrix (ECM) components, growth factor receptors, and integrins from other cells.
- pancreatic ductal adenocarcinoma
- syndecans
- proteoglycans
- tumor progression
- angiogenesis
1. Introduction
The pancreas is an organ that functions as part of the gastrointestinal system. Even though it is primarily an exocrine gland, it also has an endocrine function. The endocrine pancreas is constituted of pancreatic islets, which produce and secrete insulin and glucagon hormones (among others), to regulate blood glucose levels and glucose intake by cells. The exocrine pancreas instead is constituted by the pancreatic duct and acinar cells. It is in charge of producing enzymes such as proteases, lipases, and amylases that are released into the duodenum to support nutrient digestion. Pancreatic dysfunction can lead to digestion problems as well as dysregulation of blood glucose homeostasis due to different diseases including diabetes mellitus, chronic and acute pancreatitis, and hereditary pancreatitis. Moreover, the most relevant for patient’s survival is, without doubt, pancreatic cancer in the form of pancreatic ductal adenocarcinoma (PDAC). PDAC accounts for more than 90% of all pancreatic malignancies and is usually diagnosed at very advanced stages due to the lack of efficient screening tests present for early detection as well as due to its asymptomatic nature at early stages. Moreover, some of its symptoms, like abdominal pain, jaundice, and dark urine are not specific to this disease, which makes it even more difficult to diagnose [1]. The cause of pancreatic cancer remains unknown, but a few authors have recently expressed that some of the risk factors of developing PDAC are cigarette smoking, a diet based on a high intake of fat and meat, diabetes, alcohol abuse, and family history [2]. There are three precursors from which PDAC originates [3], namely intraductal papillary mucinous neoplasm (IPMN), mucinous cystic neoplasm (MCN) and pancreatic intraepithelial neoplasm (PanIN), the last one being the most common precursor lesion of pancreatic cancer. PanINs are non-invasive epithelial neoplasms usually located at the head of the pancreas. PanINs can be divided into PanIN-1, PanIN-2, and PanIN-3, depending on its stage. These neoplasms are considered to be the first steps before PDAC development and each one is associated with specific mutations. For example, PanIN-1 is characterized by alterations in Epithelial Growth Factor Receptor (EGFR) signaling and KRAS mutations, while PanIN-2 and -3 are characterized by the inactivation of tumor suppressor genes like CDKN2A, SMAD4, and TP53 [4].
As mentioned above, KRAS mutation is one of the first events in PanIN progression into PDAC. Constitutive activation of mutant KRas promotes plasticity of neoplastic cells and tumor development. Moreover, KRas activates signaling events such as MAPK and PI3K/Akt pathways, which regulate genes involved in cell proliferation, migration, survival/apoptosis, and metastasis [4]. These signaling pathways are normally activated through different cellular receptors such as tyrosine kinase receptors (TKRs), a receptor family which participates in a wide range of cellular processes, like cell proliferation, growth, and invasion [5]. Most of these biochemical pathways, such as cell proliferation, survival and metastasis, are possible because of the extracellular matrix (ECM). The ECM is a three-dimensional network of fibrous proteins, glycoproteins, proteoglycans, and polysaccharides with different biochemical and physical properties synthesized and secreted by stromal cells, mainly fibroblasts. Furthermore, the ECM provides structural and biochemical support for organs and tissues, epithelial cell layers as basal membrane, and individual cells as a substrate for migration [6][7]. The ECM, as well as cell–cell contact and diverse signaling molecules, serves as an information exchanger between cells forming a tissue. These interactions provide the necessary information to preserve cellular differentiation and thus create complex tissue structures. During the early stages of tumorigenesis, the ECM suffers a remodeling that supports tumor initiation and proliferation. This remodeling is caused mainly by enzymes called matrix metalloproteinases (MMP), overexpressed in most human cancers, which hydrolyze the ECM proteins [8].
Pancreatic cancer is characterized by a desmoplastic reaction, in which the deposition of abundant amounts of ECM by stromal pancreatic stellate cells (PSCs) exert biochemical and mechanical effects on PDAC cells [9]. The degree of stromal reaction predicts an aggressive phenotype, and it has been related to chemotherapeutic resistance. In fact, the accumulation of ECM components in PDAC is so high that the stroma accounts for more than 90% of the total tumor mass [10]. For this reason, the pancreatic cancer stroma has attracted the attention of many researchers in terms of targeting the ECM for the development of new PDAC treatments. However, special care should be taken to avoid the depletion of the entire ECM, as this could have dramatic consequences [10]. The degradation of the ECM components and the basal membrane implies the destruction of a physical barrier, which would allow stromal invasion by cancer cells as well as the migration of endothelial cells into the matrix, resulting in neovascularization. Moreover, during the ECM degradation, different growth factors may be released, a fact that would support the proliferation of cancer cells and tumor growth.
Another important component of the ECM which also plays a significant role in cancer cells are the proteoglycans (PGs). PGs are not only restricted to extracellular locations but are also present in cell membranes, acting as receptors to transduce extracellular signals. They are involved in functions like tissue repair and the development and maintenance of homeostatic intracellular processes. The PGs are constituted by core proteins where glycosaminoglycans (GAGs) are covalently attached. There are different types of GAGs but the most important ones are heparan sulfate (HS) and hyaluronan or hyaluronic acid (HA).
Heparan sulfate may be the most complex GAG identified until now, because of modifications and sulfations. As mentioned above, HS can be attached to a proteoglycan, forming heparan sulfate proteoglycans (HSPGs) and both are crucial for cancer initiation and progression. Importantly, HSPGs are able to modulate the interaction between growth factors and TKRs as well as the intracellular transport of extracellular vesicles carrying proteins and nucleic acids implicated in the development of cancer [11][12]. Different studies sustain the importance of these HSPGs in the development and physiology of the organisms, affecting metabolism, transport, and information transfer [13]. The major HSPGs present on the cell surface are syndecans and glypicans. Syndecans are a group of four members classified as “full-time” HSPGs because their function is restricted to the HS chains attached to the PG core protein [14].
2. Syndecan Structure and Interactions
Syndecans are type I glycoproteins encoded by the genes SDC1, SDC2, SDC3, and SDC4, which are expressed throughout the body, participating in different functions and pathways (Table 1) [15]. Syndecan structure can be divided into three differentiated domains, the ectodomain (the N-terminal polypeptide where GAGs are attached), a single transmembrane domain, and the C-terminal cytoplasmic domain (Figure 1a).
Table 1. Syndecans location and principal functions as transmembrane receptors.
|
Syndecan |
Main Location |
Function |
|
Syndecan-1 |
Epithelial and plasma cells [16] |
Cooperates with several integrins (αvβ3, αvβ5, α2β1, α3β1, and α6β4) through the core protein. Plays roles in matrix remodeling, cell adhesion and spreading, migration, and cytoskeleton rearrangements [17]. Present in breast [18][19], prostate [20], colorectal [21], and pancreatic [22][23] |
|
Syndecan-2 |
Mesenchymal cells [24] |
Important regulator of angiogenesis [25][26]. Present in some cancers such as PDAC [27] and colon cancer [28]. Regulates actin cytoskeleton organization, especially in lung cancer [29] |
|
Syndecan-3 |
Important for brain development as well as in feeding behaviors (is upregulated in the hypothalamus in response to food deprivation] [32]. Plays roles in rheumatoid arthritis disease [33], angiogenesis [34], and HIV-1 infection [35] |
|
|
Syndecan-4 |
Plays roles in cell mechanics [38] and induces focal adhesion formation [39] and cytoskeleton organization [40][41] |
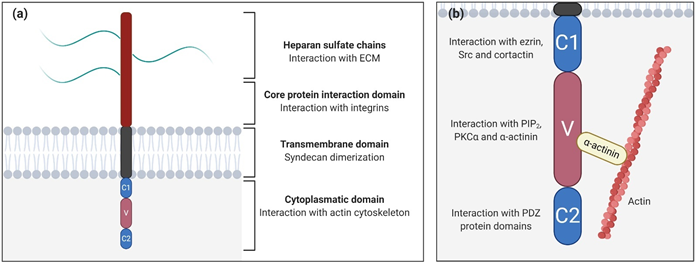
Figure 1. Syndecan structure. (a) Schematic representation of syndecan domains and their functions; (b) Close-up of the syndecan cytoplasmatic domain showing the constants (C1/C2) and the variable (V) regions and their potential interactions. The interaction between the variable region with α-actinin links syndecans to the cytoskeleton.
The ectodomain is important for cell–cell and cell–matrix interactions via attached GAGs, and introduces variances between syndecan types, unlike the other two domains that are highly conserved. In particular, heparan sulfate (HS) and chondroitin sulfate (CS) are the GAGs present in syndecan-1 and syndecan-3, while for syndecan-2 and syndecan-4, there are exclusively HS chains [42]. As GAGs are able to bind ECM molecules, syndecans, which interact with the cytoskeleton through its cytoplasmatic domain, may promote mechanical cellular responses [43]. Moreover, syndecans cooperate with specific integrins through the core protein to mediate cell adhesion [44][45]. For example, syndecan-1 cooperates with several integrins (αvβ3, αvβ5, α2β1, α3β1, and α6β4) [46][47], while syndecan-2 interacts with b1 integrins [48].
The transmembrane domain is necessary for the dimerization of core proteins into homodimers, which are essential for activating cascade signals [49]. The cytoplasmic domain is directly linked to the cytoskeleton, interacting with several kinases, and promoting different cellular functions [50]. It is divided into three subdomains (Figure 2b), two constant regions (C1 and C2) separated by a variable region (V), which is unique for every syndecan. The C1 region, the membrane-proximal domain, is associated with cytoskeletal interactions and endocytosis [51]. For example, in syndecan-2, the C1 region interacts with ezrin, an actin-associated cytoskeletal protein. Since C1 is a conserved region, the possibility exists that other syndecans could also be able to generate this interaction [52]. In syndecan-3, C1 binds to Src kinase and one of its substrates, cortactin, initiating neurite outgrowth [53]. On the other hand, the C2 region, the membrane-distal domain, interacts with PDZ binding proteins (synbindin, synectin, and syntenin) [50]. In particular, the interaction between syndecans and their adaptor protein syntenin is crucial for the biogenesis and loading of exosomes, a type of secreted vesicle involved in physiopathological processes such as cardiovascular diseases, neurodegeneration, and tumor progression. Exosomes secreted by cancer cells can contribute to tumor progression by fostering angiogenesis and the migration of tumor cells [54].
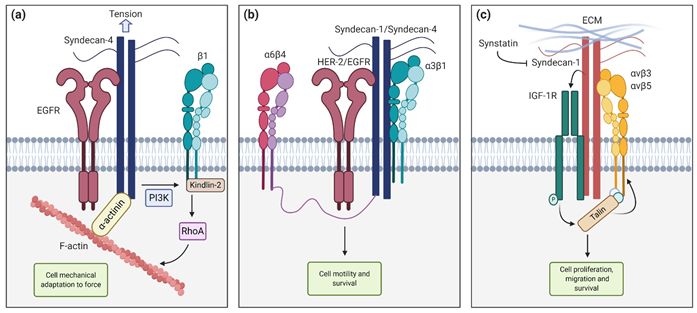
Figure 2. Syndecan functions and interactions. (a) Syndecan-4 cooperates with Epithelial Growth Factor Receptor (EGFR) and β1-integrin to tune cell mechanics in response to tension (b) Syndecan-1 and syndecan-4 interact with HER-2 and EGFR respectively, and with a6b4 and a3b1 to promote cell motility and survival (c) Syndecan-1 interaction with insulin-like growth factor-1 receptor (IGF1-R) activates talin, which in turn activates αvβ3 and αvβ5 integrins, leading to cell proliferation, migration, and survival. These interactions can be blocked with the synstatin peptide.
The V region is where most intracellular interactions between syndecans and other molecules take place. In syndecan-4, this region binds to phosphatidylinositol (4,5)-bisphosphate (PIP2) and its binding promotes the activation of PKCa [55][56]. Moreover, recent studies demonstrated that the V region motif (KKXXXKK) acts as a scaffold for PKCa, regulating its localization, activity, and stability [57]. This binding also triggers the activation of RhoA and RhoGTPases, and through syndecan-4 cooperation, it promotes focal adhesion formation [58]. The syndecan-4-PKCa interaction might be regulated by the phosphorylation of Ser183 residue . This phosphorylation decreases the affinity of syndecan-4 to PIP2 and consequently to PKCa, which in turn decreases and prevents cell migration. However, this phosphorylation promotes an alternative binding of a-actinin to the V region [59] which creates a direct linkage between syndecan-4 and the actin cytoskeleton. It has been demonstrated that interactions between the V region and a-actinin and/or PKCa are necessary for signal mechanotransduction and mechanical adaptation to force. Deletion of a-actinin and PKCa binding sites has been associated with defects in cytoskeletal organization, stress fiber assembly, and the inhibition of myofibroblast differentiation.
It is well known that syndecans interact with other membrane receptors, acting as co-receptors. They are able to associate with integrins, growth factor receptors (GFRs), as well as adhesion, invasion, angiogenic and migration promoters, and extracellular matrix glycoproteins and collagens [60]. An important aspect of this co-receptor behavior is the interaction with growth factors (GFs) and their respective GFRs. For example, Vascular Endothelial Growth Factor (VEGF) plays a key role in blood vessel formation and maintenance during development. Syndecans are believed to act as Vascular Endothelial Growth Factor Receptor (VEGFR) co-receptors by binding to its ligand and increasing its local concentration in the cell plasmatic membrane, facilitating their binding to VEGFR [61]. Indeed, in the absence of the HS chains that are present in syndecans, the VEGF would not be able to find some of its membrane receptors. Syndecans also promote ligand-receptor binding in the case of Fibroblast Growth Factor (FGF) and its receptor, where the O-sulfation pattern of the HS chains seems to be crucial for its binding capacity to FGF [62][63]
The Epithelial Growth Factor Receptor (EGFR) also interacts with the ectodomain of syndecans. In particular, Chronopoulos et al. demonstrated that when inhibiting the EGFR with Gefitinib, pancreatic stellate cells (PSC) were unable to react to external tension applied to syndecan-4, preventing adaptive cell stiffening and also force-induced PI3K activation. They concluded that syndecan-4, in cooperation with EGFR and β-1 integrin, tunes cell mechanics in response to localized tension via kindlin-2 and RhoA in a PI3K-dependent manner (Figure 2a). In another work, Wang et al. [64] described, using HaCaT and MCF10A epithelial cell lines, that syndecan-1 and syndecan-4 directly interact with HER-2 and EGFR, respectively, and also capture a3b1 integrin. This ternary complex interacts with a6b4 integrin via its cytoplasmatic link with the syndecan, leading to activation and downstream signaling that promote motility and survival (Figure 2b) [64]. Other described interactions include the association between syndecan-1, the insulin-like growth factor-1 receptor (IGF1-R) and αvβ3 or αvβ5 in both human mammary carcinoma cells and endothelial cells undergoing angiogenesis. This mechanism requires syndecan-1 clustering or engaging with matrix ligands. When captured by syndecan-1, IGF1-R suffers autophosphorylation and activation. This initiates an inside-out signaling that activates talin, which in turn promotes integrin activation. This pathway can be competitively blocked by the Synstatin (SSTN92–119), a short peptide that displaces IGF1-R and integrins from syndecan-1, preventing its activation [65][66][67] (Figure 2c).
Other examples include the cooperation between neuronal Thy-1, syndecan-4, and anb3 integrin in astrocyte cells to activate PKCa and in consequence, the RhoA pathway. This response was inhibited when SDC4 expression was silenced or a syndecan-4 mutant lacking the intracellular domain was overexpressed [68]. Syndecan-4 and a5b1 integrin also cooperates to activate PKCa in melanoma cells [69]. Moreover, the Thy-1-integrin linkage is relevant in melanoma invasion, myocyte transmigration through endothelial cells, and host defense mechanisms. In fact, the triple cooperation between Thy-1, syndecan-4, and a5b1 integrin is responsible for mediating the contractility response to mechanosignals in melanoma cells [70].
Finally, the interaction of syndecans with integrins can also be in an indirect way via intermediate receptors. For example, syndecan-2 has been described to interact with the protein tyrosine phosphatase receptor CD148 to promote Src and PI3K signaling, which in turn regulate b1 integrin-mediated adhesion processes, like angiogenesis and inflammation.
3. Syndecans in Cancer
In addition to playing many roles in development and signaling under physiological conditions, syndecans are also important in the progression of some malignancies. Their expression levels increase during cancer progression and therefore they can be considered good candidates for possible prognostic markers. For example, syndecan-1, also known as CD138, has been described to be present in multiple myeloma [71][95], breast [18,19], colorectal [21], and pancreatic carcinomas [22,23], among others. In highly metastatic breast cancer, syndecan-1 levels are higher than in those with low metastatic character, suggesting that syndecan-1 could be a remarkable biomarker for metastatic breast cancers. In particular, syndecan-1 promotes tumorigenesis via the Wnt pathway. Generation of SDC1-null transgenic mice, crossed with Wnt1-expressing mice in the mammary gland, showed that Wnt1-induced hyperplasia was reduced by 70% and that the Wnt pathway was inhibited (Table 2) [18]. Syndecan-1 also promotes cell proliferation, adhesion, and angiogenesis in mouse embryonic fibroblasts (MEFs). When co-cultured with highly invasive carcinoma cells, those MEFs that were SDC1+/+ enhanced the growth of carcinoma cells by 40% compared with SDC1−/− MEFs [72][96]. Syndecan-1 has also been used as a target for multiple myeloma treatment. In particular, Indatuximab ravtansine (BT062), an antibody-drug conjugate that binds to syndecan-1, has recently been successfully used in clinical trials in patients with relapsed multiple myeloma, stabilizing or improving disease in almost 80% of patients [73][97].
Syndecan-2 is associated with breast cancer’s metastatic ability (angiogenesis and neovascularization), morphology, and invasion index, in part by regulating RhoGTPases [74][98]. It is a survival predictor for head and neck cancer [75][99] and is also associated with colorectal cancer, in which it has implications in the migratory behavior of highly metastatic tumor cells [28].
Syndecan-3 enhances epithelial-to-mesenchymal transition (EMT) in metastatic prostate cancer, suggesting that its attenuation, and consequently its signaling pathways, could lead to a better therapeutic outcome in prostate cancer [76][100]. As other syndecans, syndecan-3 has a role in angiogenesis, with a high expression in tumoral stromal vessels [77][101].
Finally, syndecan-4 is known to be expressed in breast cancer, regulating cell adhesion and spreading and also interacting with GFRs. For example, the estradiol–estrogen receptor complex initiates the growth and progression of hormone-dependent breast cancer, and it seems that syndecan-4 participates in this pathway as a mediator factor, being activated by the Insulin-like Growth Factor Receptor (IGFR) [78][102]. In addition, the expression levels of SDC4 in human breast cancer links with the FGF2R complex formation, indicating that syndecan-4 regulates this FGF2R complex formation in human breast cancer [79][103].
3.1. Syndecans in PDAC
3.1.1. Syndecan-1
Pancreatic ductal adenocarcinoma is one of the most lethal human pathologies, mainly because of its late detection and metastatic capacity [80][3,104]. It is the only gastrointestinal pathology showing syndecan-1 upregulation [22]. This overexpression correlates with cell proliferation, differentiation, and invasion for the development of PDAC. The stroma of PDAC has increased levels of syndecan-1 compared to a normal stroma. Indeed, the shift of syndecan-1 expression from the epithelium to the stroma is a poor prognostic factor in PDAC [105]. It has also been suggested that syndecan-1 performs different functions depending on its location: epithelial syndecan-1 promotes an epithelial morphology while stromal syndecan promotes tumorigenesis [81][105]. Heparanase (HPA) is an endoglycosidase able to specifically degrade the HS chains of syndecan-1. It promotes tumor progression and metastasis by enhancing the synthesis and shedding of syndecan-1 [83]. HPA is overexpressed in pancreatic cancer [82][106] and its expression has been correlated with cancer cell invasion and lymph node metastasis in PDAC patients [83][84][107,108]. Moreover, HPA modulates the response of pancreatic cancer to radiotherapy. In particular, ionizing radiation (IR) upregulates HPA expression, promoting the invasive ability of pancreatic cancer cells in vitro and in orthotopic tumors in vivo. This could be one explanation not only for tumor resistance to radiotherapy but also for its effect in enhancing tumor dissemination. Combined treatment with an HPA inhibitor and IR attenuated spreading in orthotopic pancreatic tumors in mice [85][109]. The HPA/syndecan-1 axis promotes the upregulation of FGF2, which in turn activates the PI3K/Akt pathway and EMT in cultured pancreatic cancer cell lines [86][110]. At the same time, FGF2 also promotes syndecan-1 shedding via MMP7 in Panc-1 cells [87][111]. Moreover, syndecan-1 shedding in this cell line is enhanced by treatment with the chemotherapeutic drugs bortezomib and doxorubicin [88][112]. Increased levels of shed syndecan-1 have been reported in some cancers such as breast [19] and prostate [20] and have been correlated with poor prognosis in patients with lung cancer [89][113] and myeloma [91]. To our knowledge, there are no in vivo studies reporting the shedding of syndecan-1 in pancreatic cancer. However, the fact that HPA, FGF2, and MMP7 are all upregulated in PDAC tissue samples [90][91][110,114,115] makes it reasonable to assume that syndecan-1 shedding could also be happening during pancreatic cancer progression. This is further supported by the fact that in pancreatic cancer, epithelial syndecan-1 is produced by the epithelial cancer cells [22], but the origin of the stromal syndecan is unknown. This stromal syndecan-1 could be produced by mesenchymal cells or could be shed from epithelial cells into the tumor stroma. It is possible that the release of syndecan-1 to the stromal compartment contributes to the pancreatic malignant phenotype by binding to growth factors that support cell proliferation, as happens in breast cancer [92][93][116,117].
The KRAS mutation is the most frequent mutation in PDAC and is believed to be an initiating step for pancreatic carcinogenesis. It seems that 95% of late-stage pancreatic cancers present with a mutated and highly overexpressed KRAS. Interestingly, syndecan-1 has been described to cooperate with KRas to induce the malignant phenotype. In particular, a recent study demonstrates that syndecan-1 expression serves as a KRas effector, inducing macropinocytosis in PDAC [23]. Macropinocytosis is a type of endocytosis, which involves the non-specific intake of extracellular material, like soluble molecules, nutrients, and antigens. The study demonstrated that in low-glutamine medium, upregulated KRas cells decreased proliferative capacity. In the presence of albumin as a substitute of glutamine, cell proliferation was rescued as albumin was incorporated into the cell by macropinocytosis. SDC1 knock-out cells reduced albumin intake capacity and consequently, reduced cell proliferation in low-glutamine conditions [23]. Thus, this new study reveals that syndecan-1 plays a crucial role in macropinocytosis in KRAS-driven pancreatic cancer. In another study, the authors investigated whether there was an association between SDC1 and KRAS expression and patient survival. They found that patients carrying KRAS somatic mutations had a higher SDC1 mRNA expression than those without mutations, and that this gene signature elevated mortality[94] [118]. Both studies suggest that targeting KRAS and SDC1 in combination could improve patient outcomes[94] [23,118].
3.1.2. Syndecan-2
Syndecan-2 also plays a significant role in pancreatic cancer, working as an invasive-associated gene that, as well as syndecan-1, cooperates with KRas to induce the invasive phenotype [27]. Oliveira et al. [27] reported high syndecan-2 expression levels in various pancreatic cancer cell lines, like T3M4, Panc-1, and SU8686. When SDC2 was silenced by iRNA, migrating and invading cells were reduced significantly, although cell growth was not. In a similar way, some of the KRas/MAPK signaling pathway components, like phosphorylated Src and phosphorylated ERK, were also reduced when reducing SDC2 expression levels. Therefore, they demonstrated that syndecan-2 is an important mediator in PDAC and that it cooperates with KRas to increase malignancy and perineural invasion. The upregulation of syndecan-2 indirectly interferes with the KRas/MAPK signaling pathway enhancing the mutated KRAS gene upregulation and in consequence, the Ras protein GTP-phosphorylation. In particular, p120-GAP and RACK1 proteins compete for the binding to the syndecan-2 cytoplasmatic domain [95][119]. RACK1 plays an important role regulating the phosphorylation of Src and preventing it when Src is associated to syndecan-2. However, when p120-GAP, instead of RACK1, binds to syndecan-2, Src is phosphorylated using this binding as a switch signal [95][119]. When phosphorylated, Src interacts with several receptors, G-proteins, signal transducers, and transcription molecules, resulting in biological functions involving cell proliferation, growth, and differentiation [96][120]. The early steps for p120-GAP binding to syndecan-2 are related to the KRAS mutation. It has been demonstrated that in pancreatic cancer cell lines with wild-type KRAS, RACK1 and syndecan-2 are interacting. Alternatively, p120-GAP binds to syndecan-2 in those cell lines with mutated KRAS [27] (Figure 4). This study shows the importance of syndecan-2 in pancreatic ductal adenocarcinoma and its cooperation with oncogenic KRAS gene to aggravate this malignant phenotype. Even though syndecan-1 and -2 participate in the regulation of different processes in PDAC, both enhance KRas signaling. This fact opens a new window in PDAC treatment, as syndecan targeting could downregulate the KRas/MAPK pathway.
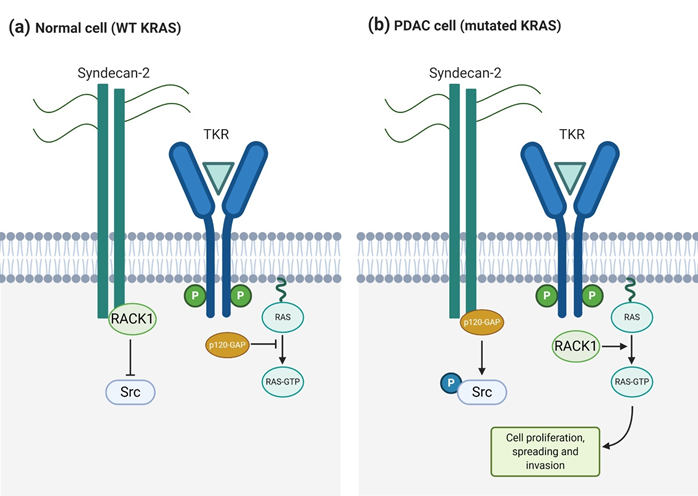
Figure 4. Syndecan-2 and KRas cooperate to induce an invasive phenotype in pancreatic cancer cells. (a) In normal cells, RACK1 binding to syndecan-2 prevents Src activation and free p120-GAP inhibits Ras signaling triggered by TKRs (b) In pancreatic ductal adenocarcinoma (PDAC) cells, p120-GAP binding to syndecan-2 activates Src and free RACK1 enhances TKR-mediated Ras activation, promoting cell proliferation, spreading, and invasion.
3.1.3. Syndecan-3 and -4
Syndecan-3 is also increased in PDAC and its expression has been positively correlated with tumor size in an orthotopic mice model [97][121]. It is associated with Midkine (MK), a type of neurotrophic factor that triggers different responses, like neurite outgrowth, neuronal survival, carcinogenesis, and tumor progression. It has been shown that the interaction between MK and syndecan-3 generates perineural invasion and poor prognosis [98][122].
Less is known about the presence and role of syndecan-4 in the pancreas. It has been detected in pancreatic islet β-cells of mice, rats, and humans, and also in the pancreatic β-cell line MIN6 [99][100] [123,124]. Importantly, syndecan-4 expression was found to be negative in other pancreatic islet cells as well as in exocrine cells, suggesting its specific role in the regulation of insulin secretion in β-cells. Moreover, SDC4 mRNA expression in β-cells is transiently upregulated by IL-1β via the Src-STAT3 pathway [99][123]. The presence and/or dysregulation of syndecan-4 has not been specifically described in PDAC and it does not seem to be relevant. However, even if not described in pancreatic duct epithelial cells, it is present in activated cultured pancreatic stellate cells (PSCs) [38]. PSCs are responsible for producing the desmoplastic stroma in which cancer cells are embedded, and therefore, for promoting PDAC progression [101][125]. Syndecan-4 has been involved in focal adhesions (FAs) formation through the binding and activation of PKCa [102][126]. The absence of syndecan-4 not only generates smaller FAs, but also shows an impaired actin-cytoskeleton and defective smooth muscle actin incorporation [103][127]. Chronopoulos et al. [38] demonstrated that applying an apical force to induce a syndecan-4 response in PSCs generates an increase in talin-1 and kindlin-2 basal accumulation. These are both focal adhesion proteins that bind the integrin cytoplasmic domain, recruit cytoskeletal, and signaling proteins involved in mechanotransduction, and enhance the integrin activation for ECM binding[104] [128]. However, this recruitment requires PI3K action. Localized force on syndecan-4 triggers PI3K activation, which enriches talin-1 and kindlin-2 concentration inducing a cell-global response, even at distal sites from the force application point. A direct link between the presence of syndecan-4 in PSCs and pancreatic cancer progression has not been described yet. However, considering the function of syndecan-4 in regulating focal adhesion formation [39] and cytoskeleton organization, [40,41], and since pancreatic cancer tissue can be several folds stiffer than its healthy counterpart [105][106][107][129–131], it would be interesting to study the role of syndecan-4 in modulating the mechanical response to the increased tissue stiffness.
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27.
- Yuan, C.; Morales-Oyarvide, V.; Babic, A.; Clish, C.B.; Kraft, P.; Bao, Y.; Qian, Z.R.; Rubinson, D.A.; Ng, K.M.; Giovannucci, E.L.; et al. Cigarette Smoking and Pancreatic Cancer Survival. J. Clin. Oncol. 2017, 35, 1822–1828.
- Goral, V. Pancreatic Cancer: Pathogenesis and Diagnosis. Asian Pacific J. Cancer Prev. 2015, 16, 5619–5624.
- Grant, T.J.; Hua, K.; Singh, A. Molecular Pathogenesis of Pancreatic Cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275.
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor-Tyrosine Kinases. Cell 2010, 141, 1117–1134.
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell, 3rd ed.; W.W Norton & Co.: New York, NY, USA, 1994.
- Hynes, R.O. The Extracellular Matrix: Not Just Pretty Fibrils. Science 2009, 326, 1216–1219.
- Klein, G.; Vellenga, E.; Fraaije, M.W.; Kamps, W.A. de Bont, E.S.J.M. The Possible Role of Matrix Metalloproteinase (MMP)-2 and MMP-9 in Cancer, e.g., Acute Leukemia. Crit. Rev. Oncol. Hematol. 2004, 50, 87–100.
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, E.; Barrett, A.S.; Hill, R.C.; Lakins, N.J.; Schlaepfer, D.; Mouw, J.K.; et al. Genotype Tunes Pancreatic Ductal Adenocarcinoma Tissue Tension to Induce Matricellular-Fibrosis and Tumor Progression. Nat. Med. 2016, 22, 497–505.
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316.
- Nagarajan, A.; Malvi, P.; Wajapeyee, N. Heparan Sulfate and Heparan Sulfate Proteoglycans in Cancer Initiation and Pro-gression. Front. Endocrinol. 2018, 9, 483.
- Christianson, H.C.; Belting, M. Heparan Sulfate Proteoglycan as a Cell-Surface Endocytosis Receptor. Matrix Biol. 2014, 35, 51–55.
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan Sulphate Proteoglycans Fine-Tune Mammalian Physiology. Nature 2007, 446, 1030–1037.
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan Sulfate Signalling in Cancer. Trends Biochem. Sci. 2014, 39, 277–288.
- Xian, X.; Gopal, S.; Couchman, J.R. Syndecans as Receptors and Organizers of the Extracellular Matrix. Cell Tissue Res. 2010, 339, 31–46.
- Saunders, S.; Jalkanen, M.; O’Farrell, S.; Bernfield, M. Molecular Cloning of Syndecan, an Integral Membrane Proteoglycan. J. Cell Biol. 1989, 108, 1547–1556.
- Beauvais, D.L.M.; Rapraeger, A.C. Syndecan-1-Mediated Cell Spreading Requires Signaling by αVβ3 Integrins in Human Breast Carcinoma Cells. Exp. Cell Res. 2003, 286, 219–232.
- Alexander, C.M.; Reichsman, F.; Hinkes, M.T.; Lincecum, J.; Becker, K.A.; Cumberledge, S. Syndecan-1 Is Required for Wnt-1-Induced Mammary Tumorigenesis in Mice. Nat. Genet. 2000, 25, 329–332.
- Malek-Hosseini, Z.; Jelodar, S.; Talei, A.; Ghaderi, A.; Doroudchi, M. Elevated Syndecan-1 Levels in the Sera of Patients with Breast Cancer Correlate with Tumor Size. Breast Cancer 2017, 24, 742–747.
- Szarvas, T.; Reis, H.; vom Dorp, F.; Tschirdewahn, S.; Niedworok, C.; Nyirady, P. Soluble Syndecan-1 (SDC1) Serum Level as an Independent Pre-Operative Predictor of Cancer-Specific Survival in Prostate Cancer. Prostate 2016, 76, 977–985.
- Wei, H.T.; Guo, E.N.; Dong, B.G.; Chen, L.S. Prognostic and Clinical Significance of Syndecan-1 in Colorectal Cancer: A Me-ta-Analysis. BMC Gastroenterol. 2015, 15, 1–8.
- Conejo, J.R.; Kleeff, J.; Koliopanos, A.; Matsuda, K.; Zhu, Z.W.; Goecke, H. Syndecan-1 Expression Is up-Regulated in Pan-creatic but Not in Other Gastrointestinal Cancers. Int. J. Cancer 2000, 88, 12–20.
- Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; Chen, Z.; Feng, N. Syndecan1 Is a Critical Mediator of Macropinocytosis in Pancreatic Cancer. Nature 2019, 568, 410–414.
- Marynen, P.; Zhang, J.; Cassiman, J.J.; Van den Berghe, H.; David, G. Partial Primary Structure of the 48- and 90-Kilodalton Core Proteins of Cell Surface-Associated Heparan Sulfate Proteoglycans of Lung Fibroblasts. Prediction of an Integral Membrane Domain and Evidence for Multiple Distinct Core Proteins at the Cell Surfa. J. Biol. Chem. 1989, 264, 7017–7024.
- Chen, E.; Hermanson, S.; Ekker, S.C. Syndecan-2 Is Essential for Angiogenic Sprouting During Zebrafish Development. Blood 2004, 103, 1710–1719.
- Noguer, O.; Villena, J.; Lorita, J.; Vilaró, S.; Reina, M. Syndecan-2 Downregulation Impairs Angiogenesis in Human Micro-vascular Endothelial Cells. Exp. Cell Res. 2009, 315, 795–808.
- De Oliveira, T.; Abiatari, I.; Raulefs, S.; Sauliunaite, D.; Erkan, M.; Kong, B.; Fries, H.; Michalski, C.W.; Kleeff, J. Syndecan-2 Promotes Perineural Invasion and Cooperates With K-Ras to Induce an Invasive Pancreatic Cancer Cell Phenotype. Mol. Cancer 2012, 11, 19.
- Vicente, C.M.; Ricci, R.; Nader, H.B.; Toma, L. Syndecan-2 Is Upregulated in Colorectal Cancer Cells Through Interactions with Extracellular Matrix Produced by Stromal Fibroblasts. BMC Cell Biol. 2013, 14, 1–14.
- Munesue, S.; Kusano, Y.; Oguri, K.; Itano, N.; Yoshitomi, Y.; Nakanishi, H.; Yamashina, I.; Okayama, M. The Role of Syndecan-2 in Regulation of Actin-Cytoskeletal Organization of Lewis Lung Carcinoma-Derived Metastatic Clones. Bio-chem. J. 2002, 363, 201–209.
- Carey, D.J.; Evans, D.M.; Stahl, R.C.; Asundi, V.K.; Conner, K.J.; Garbes, P. Molecular Cloning and Characterization of N-Syndecan, a Novel Transmembrane Heparan Sulfate Proteoglycan. J. Cell Biol. 1992, 117, 191–201.
- Gould, S.E.; Upholt, W.B.; Kosher, R.A. Syndecan 3: A Member of the Syndecan Family of Membrane-Intercalated Proteo-glycans That Is Expressed in High Amounts at the Onset of Chicken Limb Cartilage Differentiation. Proc. Natl. Acad. Sci. USA 1992, 89, 3271–3275.
- Reizes, O.; Lincecum, J.; Wang, Z.; Goldberger, O.; Huang, L.; Kaksonen, M. Transgenic Expression of Syndecan-1 Uncovers a Physiological Control of Feeding Behavior by Syndecan-3. Cell 2001, 106, 105–116.
- Patterson, A.M.; Cartwright, A.; David, G.; Fitzgeraldm, O.; Bresnihan, B.; Ashton, B.A.; Middleton, J. Differential Expression of Syndecans and Glypicans in Chronically Inflamed Synovium. Ann. Rheum. Dis. 2008, 67, 592–601.
- Arokiasamy, S.; Balderstone, M.J.M.; De Rossi, G.; Whiteford, J.R.; Syndecan-3 in Inflammation and Angiogenesis. Front. Immunol. 2020, 10, 1–7.
- De Witte, L.; Bobardt, M.; Chatterji, U.; Degeest, G.; David, G.; Geijtenbeek, T.B.H. Syndecan-3 Is a Dendritic Cell-Specific Attachment Receptor For HIV-1. Proc. Natl. Acad. Sci. USA 2007, 104, 19464–19469.
- David, G.; van der Schueren, B.; Marynen, P.; Cassiman, J.J.; van den Berghe, H. Molecular Cloning of Amphiglycan, a Nov-el Integral Membrane Heparan Sulfate Proteoglycan Expressed by Epithelial and FI-Broblastic Cells. J. Cell Biol. 1992 ,118, 961–969.
- Kojima, T.; Shworak, N.W.; Rosenberg, R.D. Molecular Cloning and Expression of Two Distinct cDNA-Encoding Heparan Sulfate Proteoglycan Core Proteins from a Rat en-Dothelial Cell Line. J. Biol. Chem. 1992, 267, 4870–4877.
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V. Syndecan-4 Tunes Cell Mechanics by Activating the Kindlin-Integrin-RhoA Pathway. Nat. Mater. 2020, 19, 669–678.
- Couchman, J.R. Syndecans: Proteoglycan Regulators of Cell-Surface Microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–937.
- Keum, E.; Kim, Y.; Kim, J.; Kwon, S.; Lim, Y.; Han, I.; Syndecan-4 Regulates Localization, Activity and Stability of Protein Kinase C-α. Biochem. J. 2004, 378, 1007–1014.
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 Associates With α-Actinin. J. Biol. Chem. 2003, 278, 7617–7623.
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavao, M.S.; Tzanakakis, G.N.; Kara-manos, N.K. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J. 2012, 279, 1177–1197.
- Yoneda, A.; Couchman, J.R. Regulation of Cytoskeletal Organization by Syndecan Transmembrane Proteoglycans. Matrix Biol. 2003, 22, 25–33.
- Whiteford, J.R.; Behrends, V.; Kirby, H.; Kusche-Gullberg, M.; Muramatsu, T.; Couchman, J.R. Syndecans Promote Integ-rin-Mediated Adhesion of Mesenchymal Cells in Two Distinct Pathways. Exp. Cell. Res. 2007, 313, 3902–3913.
- Roper, J.A.; Williamson, R.C.; Bass, M.D. Syndecan and Integrin Interactomes: Large Complexes in Small Spaces. Curr Opin. Struct Biol. 2012, 22, 583–590.
- Beauvais, D.L.M.; Burbach, B.J.; Rapraeger, A.C. The Syndecan-1 Ectodomain Regulates αVβ3 Integrin Activily in Human Mammary Carcinoma Cells. J. Cell Biol. 2004, 167, 171–181.
- McQuade, K.J.; Beauvais, D.L.M.; Burbach, B.J.; Rapraeger, A.C. Syndecan-1 Regulates αvβ5 Integrin Activity in B82L Fibro-blasts. J. Cell Sci. 2006, 119, 2445–2456.
- Whiteford, J.R.; Xian, X.; Chaussade, C., Vanhaesebroeck, B.; Nourshargh, S.; Couchman, J.R. Syndecan-2 Is a Novel Ligand for the Protein Tyrosine Phosphatase Receptor CD148. Mol. Biol. Cell 2011, 22, 3609–3624.
- Choi, S.; Lee, E.; Kwon, S.; Park, H.; Yi, J.Y.; Kim, S.; Han, I.E.; Yun, Y.; Oh, E.S.Transmembrane Domain-Induced Oligomeri-zation Is Crucial for the Functions of Syndecan-2 and Syndecan-4. J. Biol. Chem. 2005, 280, 42573–42579.
- Multhaupt, H.A.; Yoneda, A.; Whiteford, J.R.; Oh, E.S.; Lee, W.; Couchman, J.R. Syndecan Signaling: When, Where and Why? J. Physiol. Pharmacol. 2009, 60, 31–38.
- Kinnunen, A.; Kinnunen, T.; Kaksonen, M.; Nolo, R.; Panula, P.; Rauvala, H. N-syndecan and HB-GAM (Heparin-Binding Growth-Associated Molecule) associate with early axonal tracts in the rat brain. Eur. J. Neurosci. 1998, 10, 635–648.
- Granés, F.; Berndt, C.; Roy, C.; Mangeat, P.; Reina, M.; Vilaró, S. Identification of a Novel Ezrin-Binding Site in Syndecan-2 Cytoplasmic Domain. FEBS Lett. 2003, 547, 212–216.
- Kinnunen, T.; Kaksonen, M.; Saarinen, J.; Kalkkinen, N.; Peng, H.B.; Rauvala, H. Cortactin-Src Kinase Signaling Pathway Is Involved in N-Syndecan- Dependent Neurite Outgrowth. J. Biol. Chem. 1998, 273, 10702–10708.
- Zhang, H.G.; Grizzle, W.E. Exosomes: A Novel Pathway of Local and Distant Intercellular Communication That Facilitates the Growth and Metastasis of Neoplastic Lesions. Am. J. Pathol. 2014, 184, 28–41.
- Horowitz, A.; Murakami, M.; Gao, Y.; Simons, M. Phosphatidylinositol-4,5-Bisphosphate Mediates the Interaction of Syndecan-4 With Protein Kinase C. Biochemistry 1999, 38, 15871–15877.
- Whiteford, J.R.; Ko, S.; Lee, W.; Couchman, J.R. Structural and Cell Adhesion Properties of Zebrafish Syndecan-4 Are Shared with Higher Vertebrates. J. Biol. Chem. 2008, 283, 29322–29330.
- Lim, S.T.; Longley, R.L.; Couchman, J.R.; Woods, A. Direct Binding of Syndecan-4 Cytoplasmic Domain to the Catalytic Domain of Protein Kinase Cα (PKCα) Increases Focal Adhesion Localization of PKCα. J. Biol Chem. 2003, 278, 13795–13802.
- Dovas, A.; Choi, Y.; Yoneda, A.; Multhaupt, H.A.B.; Kwon, S.H.; Kang, D. Serine 34 Phosphorylation of Rho Guanine Disso-ciation Inhibitor (RhoGDIα) Links Signaling from Conventional Protein Kinase C to RhoGTPase in Cell Adhesion. J. Biol. Chem. 2010, 285, 23296–23308.
- Choi, Y.; Kim, S.; Lee, J.; Ko, S.G.; Lee, W.; Han, I.O.; Woods, A.; Oh, E.S. The Oligomeric Status of Syndecan-4 Regulates Syndecan-4 Interaction With α-Actinin. Eur. J. Cell Biol. 2008, 87, 807–815.
- Couchman, J.R. Transmembrane Signaling Proteoglycans. Annu. Rev. Cell Dev. Biol. 2010, 26, 89–114.
- Gitay-Goren, H.; Soker, S.; Vlodavsky, I.; Neufeld, G. The Binding of Vascular Endothelial Growth Factor to Its Receptors Is Dependent on Cell Surface-Associated Heparin-Like Molecules. J. Biol. Chem. 1992, 267, 6093–6098.
- Allen, B.L.; Filla, M.S.; Rapraeger, A.C. Role of Heparan Sulfate as a Tissue-Specific Regulator of FGF-4 and FGF Receptor Recognition. J. Cell. Biol. 2001, 155, 845–857.
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan Sulfate Proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–33.
- Wang, H.; Jin, H.; Rapraeger, A.C. Syndecan-1 and Syndecan-4 Capture Epidermal Growth Factor Receptor Family Members and the α3β1 Integrin via Binding Sites in Their Ecto-Domains: Novel Synstatins Prevent Kinase Capture and In-hibitα6β4-Integrindependent Epithelial Cell Motility. J. Biol. Chem. 2015, 290, 26103–26113.
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 Regulates αvβ3 and αvβ5 Integrin Activation During Angiogenesis and Is Blocked by Synstatin, a Novel Peptide Inhibitor. J. Exp. Med. 2009, 206, 691–705.
- Beauvais, D.L.M.; Rapraeger, A.C. Syndecan-1 Couples the Insulin-Like Growth Factor-1 Receptor to Inside-Out Integrin Activation. J. Cell Sci. 2010, 123, 3796–3807.
- Rapraeger, A.C. Synstatin: A Selective Inhibitor of the Syndecan-1-Coupled IGF1R-αvβ3 Integrin Complex in Tumorigene-sis and Angiogenesis. FEBS J. 2013, 280, 2207–2215.
- Avalos, A.M.; Valdivia, A.D.; Muñoz, N.; Herrera-Molina, R.; Tapia, J.C.; Lavandero, S. Neuronal Thy-1 Induces Astrocyte Adhesion by Engaging Syndecan-4 in a Cooperative Interaction With αVβ3 Integrin That Activates PKCα and RhoA. J. Cell Sci. 2009, 122, 3462–3471.
- Mostafavi-Pour, Z.; Askari, J.A.; Parkinson, S.J.; Parker, P.J.; Ng, T.T.; Humphries, M.J. Integrin-Specific Signaling Pathways Controlling Focal Adhesion Formation and Cell Migration. J. Cell Biol. 2003, 161, 155–167.
- Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic Catch of a Thy-1-α5 β1 + Syndecan-4 Trimolecular Complex. Nat. Commun. 2014, 5.
- Sanderson, R.D.; Yang, Y.; Syndecan-1: A Dynamic Regulator of the Myeloma Microenvironment. Clin Exp Metastasis 2008, 25, 149–159.Fiore, V.F.; Ju, L.; Chen, Y.; Zhu, C.; Barker, T.H. Dynamic Catch of a Thy-1-α5 β1 + Syndecan-4 Trimolecular Complex. Nat. Commun. 2014, 5.
- Maeda, T.; Alexander, C.M.; Friedl, A. Induction of Syndecan-1 Expression in Stromal Fibroblasts Promotes Proliferation of Human Breast Cancer Cells. Cancer Res. 2004, 64, 612–621.
- Jagannath, S.; Heffner, L.T.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Indatuximab Ravtansine (BT062) Monotherapy in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin Lymphoma Myeloma Leuk. 2019, 19, 372–380.
- Lim, H.C.; Couchman, J.R. Syndecan-2 Regulation of Morphology in Breast Carcinoma Cells Is Dependent on RhoGTPases. Biochim. Biophys. Acta—Gen. Subj. 2014, 1840, 2482–2490.
- Farnedi, A.; Rossi, S.; Bertani, N.; Gulli, M.; Silini, E.M.; Mucignat, M.T.; Poli, T.; Sesenna, E.; Lanfranco, D.; Montebugnoli, L.; et al. Proteoglycan-Based Diversification of Disease Outcome in Head and Neck Cancer Patients Identifies NG2/CSPG4 and Syndecan-2 as Unique Relapse and Overall Survival Predicting Factors. BMC Cancer. 2015, 15, 1–19.
- Diamantopoulou, Z.; Kitsou, P.; Menashi, S.; Courty, J.; Katsoris, P. Loss of Receptor Protein Tyrosine Phosphatase β/ζ (RPTPβ/ζ) Promotes Prostate Cancer Metastasis. J. Biol. Chem. 2012, 287, 40339–40349.
- Roskams, T.; De Vos, R.; David, G.; Van Damme, B.; Desmet, V. Heparan Sulphate Proteoglycan Expression in Human Pri-mary Liver Tumours. J Pathol. 1998, 185, 290–297.
- Tsonis, A.I.; Afratis, N.; Gialeli, C.; Ellina, M.I.; Piperigkou, Z.; Skandalis, S.S. Evaluation of the Coordinated Actions of Estrogen Receptors with Epidermal Growth Factor Receptor and Insulin-Like Growth Factor Receptor in the Expression of Cell Surface Heparan Sulfate Proteoglycans and Cell Motility in Breast Cancer Cells. FEBS J. 2013, 280, 2248–2259.
- Mundhenke, C.; Meyer, K.; Drew, S.; Friedl, A. Heparan Sulfate Proteoglycans as Regulators of Fibroblast Growth Factor-2 Receptor Binding in Breast Carcinomas. Am. J. Pathol. 2002, 160, 185–194.
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081.
- Juuti, A.; Nordling, S.; Lundin, J.; Louhimo, J.; Haglund, C. Syndecan-1 Expression—A Novel Prognostic Marker in Pancre-atic Cancer. Oncology 200, 68, 97–106.
- Koliopanos, A.; Friess, H.; Kleeff, J.; Shi, X.; Liao, Q.; Pecker, I. Heparanase Expression in Primary and Metastatic Pancreatic Cancer. Cancer Res. 2001, 61, 4655–4659.
- Rohloff, J.; Zinke, J.; Schoppmeyer, K.; Tannapfel, A.; Witzigmann, H.; Mössner, J. Heparanase Expression Is a Prognostic Indicator for Postoperative Survival in Pancreatic Adenocarcinoma. Br. J. Cancer. 2002, 86, 1270–1275.
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Drebber, U.; Baldus, S.E. High Expression of Heparanase Is Signif-icantly Associated with Dedifferentiation and Lymph Node Metastasis in Patients With Pancreatic Ductal Adenocarcinomas and Correlated to PDGFA and via HIF1a to HB-EGF and bFGF. J. Gastrointest. Surg. 2008, 12, 1674–1681.
- Meirovitz, A.; Hermano, E.; Lerner, I.; Zcharia, E.; Pisano, C.; Peretz, T.; Elkin, M. Role of Heparanase in Radiation-Enhanced Invasiveness of Pancreatic Carcinoma. Cancer Res. 2011, 71, 2772–2780.
- Chen, X.; Zhao, H.; Chen, C.; Li, J.; He, J.; Fu, X. The HPA/SDC1 Axis Promotes Invasion and Metastasis of Pancreatic Cancer Cells by Activating EMT via FGF2 Upregulation. Oncol. Lett. 2019, 19, 211–220.
- Ding, K.; Lopez-Burks, M.; Sánchez-Duran, J.A.; Korc, M.; Lander, A.D. Growth Factor-Induced Shedding of Syndecan-1 Confers Glypican-1 Dependence on Mitogenic Responses of Cancer Cells. J. Cell Biol. 2005, 171, 729–738.
- Ramani, V.C.; Sanderson, R.D. Chemotherapy Stimulates Syndecan-1 Shedding: A Potentially Negative Effect of Treatment That May Promote Tumor Relapse. Matrix Biol. 2014, 35, 215–222.
- Joensuu, H.; Anttonen, A.; Eriksson, M.; Mäkitaro, R.; Alfthan, H.; Kinnula, V.; Sirpa, L. Soluble Syndecan-1 and Serum Basic Fibroblast Growth Factor Are New Prognostic Factors in Lung Cancer. Cancer Res. 2002, 62, 5210–5217.
- Yamamoto, H.; Itoh, F.; Iku, S.; Adachi, Y.; Fukushima, H.; Sasaki, S.; Mukaiya, M.; Hirata, K.; Imai, K. Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Human Pancreatic Adenocarcinomas: Clinicopathologic and Prognostic Significance of Matrilysin Expression. J. Clin. Oncol. 2001, 19, 1118–1127.
- Crawford, H.C.; Scoggins, C.R.; Washington, M.K.; Matrisian, L.M.; Leach, S.D. Matrix Metalloproteinase-7 Is Expressed by Pancreatic Cancer Precursors and Regulates Acinar-to-Ductal Metaplasia in Exocrine Pancreas. J. Clin. Invest. 2002, 109, 1437–1444.
- Su, G.; Blaine, S.A.; Qiao, D.; Friedl, A. Shedding of Syndecan-1 by Stromal Fibroblasts Stimulates Human Breast Cancer Cell Proliferation via FGF2 Activation. J. Biol. Chem. 2007, 282, 14906–14915.
- Nikolova, V.; Koo, C.-Y.; Ibrahim, S.A.; Wang, Z.; Spillmann, D.; Dreier, R. Differential Roles for Membrane-Bound and Soluble Syndecan-1 (CD138) in Breast Cancer Progression. Carcinogenesis 2009, 30, 397—407.
- Wu, Y.; Huang, H.; Fervers, B.; Lu, L. Syndecan-1 and KRAS Gene Expression Signature Associates with Patient Survival in Pancreatic Cancer. Pancreas 2020, 49, 1187–1194.
- Huang, J.W.; Chen, C.L.; Chuang, N.N. P120-GAP Associated with Syndecan-2 to Function as an Active Switch Signal for Src Upon Transformation with Oncogenic Ras. Biochem. Biophys. Res. Commun. 2005, 329, 855–862.
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The Role of Src in Solid Tumors. Oncologist 2009, 14, 667.
- Yao, J.; Zhang, L.L.; Huang, X.M.; Li, W.Y.; Gao, S.G. Pleiotrophin and N-Syndecan Promote Perineural Invasion and Tumor Progression in an Orthotopic Mouse Model of Pancreatic Cancer. World J. Gastroenterol. 2017, 23, 3907–3914.
- Yao, J.; Li, W.Y.; Li, S.G.; Feng, X.S.; Gao, S.G. Midkine Promotes Perineural Invasion in Human Pancreatic Cancer. World J. Gastroenterol. 2014, 20, 3018–3024.
- Brioudes, E.; Alibashe-Ahmed, M.; Lavallard, V.; Berney, T.; Bosco, D. Syndecan-4 Is Regulated by IL-1β in β-Cells and Hu-man Islets. Mol. Cell Endocrinol. 2020, 510, 110815.
- Cheng, J.Y.C.; Whitelock, J.; Poole-Warren, L. Syndecan-4 Is Associated with Beta-Cells in the Pancreas and the min6 Be-ta-Cell Line. Histochem. Cell Biol. 2012, 138, 933–944.
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J. A.; et al. Desmoplastic Reaction in Pancreatic Cancer: Role of Pancreatic Stellate Cells. Pancreas 2004, 29, 179–187.
- Fogh, B.S.; Multhaupt, H.A.B.; Couchman, J.R. Protein Kinase C, Focal Adhesions and the Regulation of Cell Migration. J. Histochem. Cytochem. 2014, 62, 172–184.
- Gopal, S.; Bober, A.; Whiteford, J.R.; Multhaupt, H.A.B.; Yoneda, A.; Couchman, J.R. Heparan Sulfate Chain Valency Con-trols Syndecan-4 Function in Cell Adhesion. J Biol. Chem. 2010, 285, 14247–14258.
- Calderwood, D.A.; Campbell, I.S.; Critchley, D.R. Talins and Kindlins: Partners in Integrin-Mediated Adhesion. Nat. Rev. Mol. 2013, 14, 503–517.
- Itoh, Y.; Takehara, Y.; Kawase, T.; Terashima, K.; Ohkawa, Y.; Hirose, Y.; Koda, A.; Hyodo, N.; Ushio, T.; Hirai, Y.; et al. Fea-sibility of Magnetic Resonance Elastography for the Pancreas at 3T. J. Magn. Reson. Imaging 2016, 43, 384–390.
- Rubiano, A.; Delitto, D.; Han, S.; Gerber, M.; Galitz, C.; Trevino, J.; Thomas, R.M.; Hughes, S.J.; Simmons, C.S. Viscoelastic Properties of Human Pancreatic Tumors and in Vitro Constructs to Mimic Mechanical Properties. Acta Biomater. 2017, 67, 331–340.
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Hernandez, A.D.R. Matrix Stiffness Induces Epithelial—Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e342–e352.
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Hernandez, A.D.R. Matrix Stiffness Induces Epithelial—Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e342–e352.