Epithelial–mesenchymal transition (EMT) is a complex cellular program known to be a crucial driver in the context of embryonic development, wound healing and tumour progression.
- epithelial-mesenchymal transition
- fibrosis
- TGF-β
- autoimmune diseases
- inflammation
1. Introduction
Epithelial–mesenchymal transition (EMT) is a complex cellular program known to be a crucial driver in the context of embryonic development, wound healing and tumour progression. EMT involves intense changes in the morphology and behaviour of epithelial cells. Indeed, not only do they lose their original phenotypic and functional features, but they acquire the capacity to degrade the basement membrane, migrating through the extracellular matrix to populate different territories either during embryonic development or cancer progression, or to adopt a profibrotic myofibroblast nature in the tissue interstitial space[1][2][3][4][5] [1,2,3,4,5]. This process is characterized by a dramatic change in the expression of specific epithelial proteins, such as E-cadherin and zonula occludens-1 (ZO-1), followed by a markedly increased expression of some mesenchymal markers, including α-smooth muscle actin (α-SMA), vimentin, and laminin[6] [6] (Figure 1).
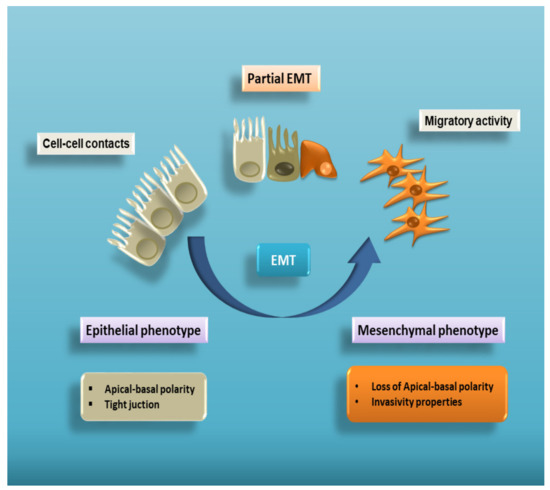
Figure 1. Cell plasticity in EMT. EMT is a multistep process allowing epithelial cells to lose their apical–basal polarity and disassemble epithelial cell–cell contacts. Through a complex cellular and molecular program, these cells progressively acquire mesenchymal features that include cytoskeleton reorganization and a proteolytic capacity favouring cell motility. Therefore, EMT proceeds through multiple partial intermediate states, collectively known as the partial EMT. A partial EMT takes place physiologically during wound healing and leads to an intermediate phenotype that presents some epithelial features, but also characteristics of mesenchymal cells.
Although EMT is involved in various biological processes, such as embryogenesis and tissue repair, it plays a key cellular role in the development of fibrotic disorders of mature organs, which are often the outcome of pathological chronic disease. Indeed, in animal models, inhibition of EMT has proven beneficial in attenuating the progression of tissue fibrosis[7][8] [7,8].
2. TGF-β Activation and Signalling
The TGF-β signalling pathway is known to act physiologically as a regulator of embryogenesis, adaptive and innate immunity, cancer development, inflammation, and fibrosis[9] [36]. However, perturbations in TGF-β signalling determine TGF-β switching, accelerating the development or progression of cancer, congenital defects and fibrosis in chronic inflammatory autoimmune conditions[10][11] [35,37]. During the course of chronic autoimmune diseases, in fact, the activation of fibrogenic mediators produced by inflammatory and epithelial cells was observed. In this context, TGF-β1 emerged as a crucial factor and fibrogenesis, in the majority of the organ where it occurs, is characterized by persistent inflammation, altered interactions between epithelial and mesenchymal cells, and fibroblasts proliferation[12][13] [38,39].
One of the hallmarks of excessive pathological fibrogenesis is the acquisition by fibroblasts of a highly activated myofibroblasts phenotype characterized by contractile cells that express α-SMA, exhibiting dysregulation of the ECM composition and structure. The recruitment of additional immune cells into the fibrotic tissue amplifies the fibrotic response, because these recruited cells can also secrete a variety of chemokines, cytokines, and growth factors responsible for the differentiation of other myofibroblasts and stimulation of ECM deposition[14][15] [40,41]. Great attention is paid in the recent literature to the immunological functions of TGF-β; indeed, it exerts a broad anti-inflammatory activity and is considered to be an immunosuppressive agent[16][17] [42,43]. Complete gene silencing of TGF-β1 in mice could lead to death owing to a multi-organ inflammatory syndrome[18][19] [44,45]. However, not all TGF-β effects are suppressive; indeed, TGF-β has the capacity to induce either immunosuppressive or inflammatory events, depending on the molecular situation. For example, the Th-17 effector cells are able to acquire their pathogenic function through the mediation of TGF-β and IL-6[20] [46] and this has been linked to hyperactivation of the immune system caused by the release of inflammatory factors that could ultimately lead to autoimmunity conditions.
Traditionally, TGF-β is a multifunctional cytokine produced by immune cells. TGF-β1 is the prevalent isoform normally found in plasma[21] [47] and ubiquitously bound to ECM proteins in mammalian tissues[21] [47]. Interestingly, large amounts of TGF-β1 are produced by platelets and bones[22][23] [48,49]. Unlike other cytokines, TGF-β1 is secreted in a latent form that consists of its dimeric pro-peptide [known as the latency-associated peptide (LAP)], and a latent TGF-β-binding protein (LTBP). This tripartite complex of TGF-β, LAP, and LTBP is called the large latent complex (LLC)[24] [50]. TGF-β activation requires proteolysis by plasmin (a factor of the fibrinolytic system) and other proteases. At least in some cases, plasmin-mediated activation occurs on the cell membrane. For instance, the mannose-6-phosphate/insulin-like growth factor II receptor (M6P/IGFII-R) binds LAP-TGF-β on the surface of monocytes and, acting with the urokinase receptor and plasminogen to generate plasmin, determines the activation of the latent TGF-β[25] [51] However, as reported in the literature, other molecules such as platelets expressing thrombospondin 1 (TSP-1), and αvβ6 integrin, an epithelial-cell membrane protein, can also bind and activate LAP-TGF-β[26][27] [52,53]. Interestingly, the matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9) have been implicated as activators of TGF-β following the involvement of the CD44 hyaluronan receptor[28] [54]. This mechanism, based on the interactions of CD44, MMPs, and TGF-β on the cell membrane, seems to affect cancer cell motility, invasion, and metastasis[29] [55].
3. TGF-β Mediates EMT-Dependent Fibrosis
The induction of EMT by TGF-β was first recognized in cell culture. Upon TGF-β treatment, epithelial cells changed from a cuboidal to an elongated spindle shape, and showed a decreased expression of epithelial markers and enhanced expression of the mesenchymal markers fibronectin and vimentin[30] [56]. These changes were accompanied by an increased motility. Consistent with their binding to the same receptor complexes, TGF-β1, TGF-β2, and TGF-β3 share the capacity to induce EMT in epithelial cells[31][32] [57,58]. Subsequent studies have demonstrated an involvement of TGF-β and TGF-β-related proteins in EMT during normal development and in pathological processes. Fibrosis is characterized by an excessive accumulation of extracellular matrix in the affected tissue; the fibrotic process occurs in various organs and although it can have multiple causes, organ fibrosis typically results from chronic persistent inflammation induced by numerous different stimuli[33] [59]. TGF-β1 is a most important factor involved in the activation of tissue fibrosis, as demonstrated in liver[34] [60], in kidney[35] [32], and in lung[36] [61]. TGF-β1 may generate signals that trigger the EMT program in epithelial cells whose morphology is transformed to that of mesenchymal cells. They produce extracellular matrix components, enriched with fibroblasts in the fibrotic wound, and thus participate in EMT-dependent fibrotic tissue formation[11][37] [37,62]. When excessive fibrotic production occurs, owing to its profibrotic effects, TGF-β contributes to an excessive tissue fibrosis, leading to organ dysfunction and failure[38] [63]. Indeed, inflammation is recognized as the earliest stage of fibrosis, and emerging data point to the potential role of chronic long-term inflammation as responsible for the development of fibrogenesis[39][40] [64,65].
During development and in the context of different morphogenetic and/or pathological events such as carcinogenesis, and fibrosis, TGF-β exerts a primary role, inducing various effects depending on the specific cellular context and specific isoforms involved[41] [66]. The first evidence of a role of TGF-β1 in EMT was derived from studies conducted in normal mammary epithelial cells[42] [67]. Since these preliminary studies, TGF-β1 has been shown to mediate EMT in numerous different epithelial cells derived from the lung, liver, lens, or kidney[43][44] [68,69]. TGF-β is considered to be the prototypical cytokine for the induction of EMT, whereas the effects of other molecules reveal a cell context-dependent mechanism of action[44] [69]. Major evidence of the action of TGF-β on triggering EMT also derives from studies conducted in vivo, especially by means of modulating the TGF-β-dependent Smad pathway in animal models. Knockout of the Smad3 gene or the employment of Smad7, which works as an antagonist of TGF-β signalling, or of bone morphogenetic protein-7 (BMP-7) acting in a Smad-dependent manner, offsets EMT in mice [45][46][70,71]. It was also shown to slow and reverse uncontrolled fibrosis processes in renal and lens epithelia[47][48] [18,72]. During pulmonary fibrosis, potential sources of lung fibroblasts include the proliferation of resident lung interstitial fibroblasts, progenitor cell differentiation, as well as the transition of epithelial cells to a fibroblast phenotype through the EMT process[4] [4]. Confirming this hypothesis, several authors clearly demonstrated that, in vivo, approximately one third of the lung fibroblasts able to release the fibrotic protein S100A4 derive from the lung epithelium after the administration of bleomycin, widely used as an inducer of pulmonary fibrosis in animal models[49][50] [73,74].
The direct involvement of TGF-β in the EMT-dependent differentiation of lung fibroblasts, in vivo, was recently demonstrated by the detection of increased active TGF-β1 in transgenic mouse permanently β-galactosidase stained alveolar epithelial cells. In this model, X-gal-positive cells underwent phenotypical changes of EMT, expressing the myofibroblast marker α-SMA, while most of the cells identified as X-gal-positive fibroblasts expressed vimentin, indicating an epithelial origin[51] [75].
Reference (Editors will rearrange the references after the entry is submitted)
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890.
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449.
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437.
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784.
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Investig. 2009, 119, 1417–1419.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2014, 15, 178–196.
- López-Novoa, J.M.; Nieto, M.A. Inflammation and EMT: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314.
- O’Connor, J.W.; Gomez, E.W. Biomechanics of TGFβ-induced epithelial-mesenchymal transition: Implications for fibrosis and cancer. Clin. Trans. Med. 2014, 15, 3–23.
- Li, M.; Luan, F.; Zhao, Y.; Hao, H.; Zhou, Y.; Han, W.; Fu, X. Epithelial-mesenchymal transition: An emerging target in tissue fibrosis. Exp. Biol. Med. 2016, 241, 1–13.
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Neilson Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350.
- Radisky, D.C.; Kenny, P.A.; Bissell, M.J. Fibrosis and cancer: Do myofibroblasts come also from epithelial cells via EMT? J. Cell Biochem. 2007, 101, 830–839.
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040.
- Jiang, H.; Shen, J.; Ran, Z. Epithelial–mesenchymal transition in Crohn’s disease. Mucosal Immunol. 2018, 11, 294–303.
- Jinde, K.; Nikolic-Paterson, D.J.; Huang, X.R.; Sakai, H.; Kurokawa, K.; Atkins, R.C.; Lan, H.Y. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am. J. Kidney Dis. 2001, 38, 761–769.
- Chilosi, M.; Poletti, V.; Zamò, A.; Lestani, M.; Montagna, L.; Piccoli, P.; Pedron, S.; Bertaso, M.; Scarpa, A.; Murer, B.; et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am. J. Pathol. 2003, 162, 1495–1502.
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347.
- Sisto, M.; Lisi, S.; Ribatti, D. The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands. Histochem. Cell Biol. 2018, 150, 133–147.
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968.
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24.
- Yoshimatsu, Y.; Watabe, T. Roles of TGF-β signals in endothelial-mesenchymal transition during cardiac fibrosis. Int. J. Inflam. 2011, 2011, 724080.
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222.
- Strutz, F.M. EMT and proteinuria as progression factors. Kidney Int. 2009, 75, 475–481.
- Fragiadaki, M.; Mason, R.M. Epithelial-mesenchymal transition in renal fibrosis-evidence for and against. Int. J. Exp. Pathol. 2011, 92, 143–150.
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell. Dev. Biol. 2010, 27, 347–376.
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Ribatti, D.; Lisi, S. TGFβ1-Smad canonical and -Erk non-canonical pathways participate in interleukin-17-induced epithelial–mesenchymal transition in Sjögren’s syndrome. Lab. Investig. 2020, 100, 824–836.
- Zvaifler, N.J. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res. Ther. 2006, 8, 210.
- Chen, S.Y. Does epithelial-mesenchymal transition happen in rheumatoid joints? Eur. J. Rheumatol. 2014, 1, 86–87.
- Fintha, A.; Gasparics, Á.; Rosivall, L.; Sebe, A. Therapeutic Targeting of Fibrotic Epithelial-Mesenchymal Transition-An Outstanding Challenge. Front. Pharmacol. 2019, 10, 388.
- Lotz, M.; Kekow, J.; Carson, D.A. Transforming growth factor-beta and cellular immune responses in synovial fluids. J. Immunol. 1990, 144, 4189–4194.
- Flier, S.N.; Tanjore, H.; Kokkotoum, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212.
- Rastaldi, M.P. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J. Nephrol. 2006, 19, 407–412.
- Carew, R.M.; Wang, B.; Kantharidis, P. The role of EMT in renal fibrosis. Cell Tissue Res. 2012, 347, 103–116.
- Leehan, K.M.; Pezant, N.P.; Rasmussen, A.; Grundahl, K.; Moore, J.S.; Radfar, L.; Lewis, D.M.; Stone, D.U.; Lessard, C.J.; Rhodus, N.L.; et al. Minor salivary gland fibrosis in Sjögren’s syndrome is elevated, associated with focus score and not solely a consequence of aging. Clin. Exp. Rheumatol. 2018, 112, 80–88.
- Ficarra, B.J. Submandibular salivary gland fibrosis. J. Med. 1996, 27, 103–113.
- Sisto, M.; Lorusso, L.; Ingravallo, G.; Tamma, R.; Ribatti, D.; Lisi, S. The TGF-β1 Signaling Pathway as an Attractive Target in the Fibrosis Pathogenesis of Sjögren’s Syndrome. Mediat. Inflamm. 2018, 2018, 1965935.
- Yoshimura, A.; Wakabayashi, Y.; Mori, T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J. Biochem. 2010, 147, 781–792.
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202.
- Schultz-Cherry, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338.
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 signaling and tissue fibrosis. Perspect. Biol. 2018, 10, a022293.
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The Fibroblast awakens. Circ. Res. 2016, 118, 1021–1040.
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of gammadelta T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642.
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146.
- Rubtsov, Y.P.; Rudensky, A.Y. TGF beta signalling in control of T-cell-mediated self-reactivity. Nat. Rev. Immunol. 2007, 7, 443–453.
- Kallapur, S.; Shull, M.; Doetchman, T. Phenotypes of TGF beta knockout mice. In Cytokine Knockouts; Durum, S.K., Muegge, K., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 335–368.
- Kulkarni, A.B.; Letterio, J.J. The transforming growth factor beta-1 mouse: The phenotype and its implications for TGFbeta1 function. In Cytokine Knockouts; Durum, S.K., Muegge, K., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 369–400.
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397.
- Howe, P.H. Transforming growth factor β. In The Cytokine Handbook, 4th ed.; Thompson, A.W., Lotze, M.T., Eds.; Academic Press: San Diego, CA, USA, 2003; pp. 1119–1152.
- Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latent TGF-beta binding proteins: Extracellular matrix association and roles in TGF-beta activation. Crit. Rev. Clin. Lab. Sci. 2004, 4, 233–264.
- Fox, S.W.; Lovibond, A.C. Current insights into the role of transforming growth factor-beta in bone resorption. Mol. Cell. Endocrinol. 2005, 243, 19–26.
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176.
- Godar, S.; Horejsi, V.; Weidle, U.H.; Binder, B.R.; Hansmann, C.; Stockinger, H. M6P/IGFII–receptor complexes urokinase receptor and plasminogen for activation of transforming growth factor-beta1. Eur. J. Immunol. 1999, 29, 1004–1013.
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328.
- Schultz-Cherry, S.; Ribeiro, S.; Gentry, L.; Murphy-Ullrich, J.E. Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J. Biol. Chem. 1994, 269, 26775–26782.
- Krstic, J.; Santibanez, J.F. Transforming growth factor-beta and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014, 21, 521754.
- Yu, Q.; Stamenkovic, I. Transforming growth factor-beta facilitates breast carcinoma metastasis by promoting tumor cell survival. Clin. Exp. Metastasis 2004, 2, 235–242.
- Todorovic, V.; Jurukovski, V.; Chen, Y.; Fontana, L.; Dabovic, B.; Rifkin, D.B. Latent TGF-beta binding proteins. Int. J. Biochem. Cell. Biol. 2005, 37, 38–41.
- Piek, E.; Moustakas, A.; Kurisaki, A.; Heldin, C.H.; ten Dijke, P. TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 1999, 112, 4557–4568.
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell. 2005, 16, 1987–2002.
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210.
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dewidar, B.; Giannelli, G.; Ten Dijke, P. IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232.
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. 2007, 293, L525–L534.
- Zeisberg, M.; Kalluri, R. Fibroblasts emerge via epithelial-mesenchymal transition in chronic kidney fibrosis. Front. Biosci. 2008, 13, 6991–6998.
- Kanzler, S.; Lohse, A.W.; Keil, A.; Henninger, J.; Dienes, H.P.; Schirmacher, P.; Rose-John, S.; zum Büschenfelde, K.H.; Blessing, M. TGF-beta1 in liver fibrosis: An inducible transgenic mouse model to study liver fibrogenesis. Am. J. Physiol. 1999, 276, G1059–G1068.
- Kaimori, A.; Potter, J.; Kaimori, J.; Wang, C.; Mezey, E.; Koteish, A. Transforming growth factor-β1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J. Biol. Chem. 2007, 282, 22089–22101.
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF beta and EGFR signaling. PLoS ONE 2013, 8, e54001.
- Wick, G.; Backovic, A.; Rabensteiner, E.; Plank, N.; Schwentner, C.; Sgonc, R. The immunology of fibrosis: Innate and adaptive responses. Trends Immunol. 2010, 31, 110–119.
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036.
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009, 5, 1145–1168.
- Kriz, W.; Kaissling, B.; Le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Investig. 2011, 121, 468–474.
- Meng, F.; Li, J.; Yang, X.; Yuan, X.; Tang, X. Role of Smad3 signalling in the epithelial-mesenchymal transition of the lens epithelium following injury. Int. J. Mol. Med. 2018, 42, 851–860.
- Itoh, F.; Asao, H.; Sugamura, K.; Heldin, C.H.; ten Dijke, P.; Itoh, S. Promoting bone morphogenetic protein signalling through negative regulation of inhibitory Smads. EMBO J. 2001, 20, 4132–4142.
- Hales, A.M.; Schulz, M.W.; Chamberlain, C.G.; McAvoy, J.W. TGF-beta 1 induces lens cells to accumulate alpha-smooth muscle actin, a marker for subcapsular cataracts. Curr. Eye Res. 1994, 13, 885–890.
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care. Med. 2009, 180, 657–665.
- Chen, L.J.; Ye, H.; Zhang, Q.; Li, F.Z.; Song, L.J.; Yang, J.; Mu, Q.; Rao, S.S.; Cai, P.C.; Xiang, F.; et al. Bleomycin induced epithelial-mesenchymal transition (EMT) in pleural mesothelial cells. Toxicol. Appl. Pharmacol. 2015, 283, 75–82.
- Wu, Z.; Yang, L.; Cai, L.; Zhang, M.; Cheng, X.; Yang, X.; Xu, J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir. Res. 2007, 8, 1.