Necroptosis is a caspase independent form of regulated cell death, investigated as a novel therapeutic strategy to eradicate apoptosis resistant cancer cells. The process can be triggered by a variety of stimuli and is controlled by the activation of RIP kinases family as well as MLKL. The well-studied executor, RIPK1, is able to modulate key cellular events through the interaction with several proteins, acting as strategic crossroads of several molecular pathways. Little evidence is reported about the involvement of RIPK1 in tumorigenesis. Of particular interest is its contradictory role in cancer and its function in cell fate control. Targeting necroptosis might be a novel therapeutic intervention strategy in anticancer therapies as pharmacologically controllable event.
- necroptosis
- cancer
- RIPK1
- cell death
1. Introduction
Proliferation, differentiation, and cell death are physiological events responsible for the maintenance of cellular homeostasis [1]. Each cell has a well-defined life cycle at the end of which it dies naturally [2]. Cell death can occur in two different modalities, one following a passive, uncontrolled or accidental process, the other one through an active and regulated process [3].
Cells die not only as a physiological phenomenon but also when exposed to chemical, physical, and mechanical insults and, based on the severity of the trauma, we distinguish accidental cell death (ACD), a catastrophic and extreme event with consequent disruption of cellular structure since it is completely unprogrammed, with regulated cell death (RCD), which is a controlled process, depending on activation of signaling cascades and hence can be genetically and pharmacologically controlled [4][5][6][4,5,6].
When RCD occurs in a physiological scenario with the preservation of development and tissue homeostasis, it is known as programmed cell death (PCD) [3][5][3,5] (Figure 1).
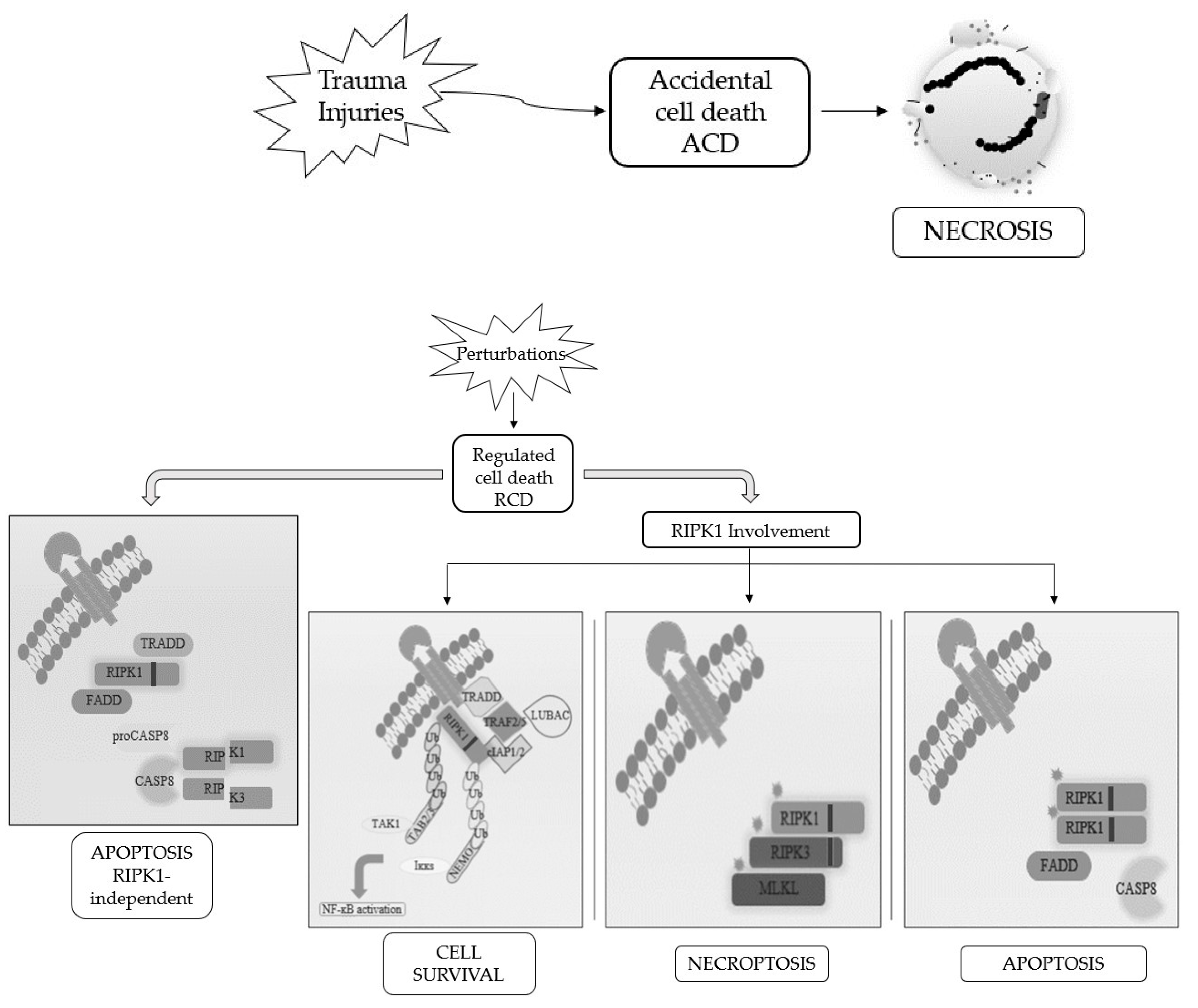
Figure 1. Schematic representation of the best-known cell death modalities with different molecular features.
Apoptosis and necrosis were the first and so the best known two modalities of cell death discovered. Historically, apoptosis has been associated with non-immunogenic, regulated, and programmed forms of death, while necrosis has been defined as accidental and uncontrolled [6]. Apoptosis is a defense mechanism that occurs physiologically as a result of extracellular or intracellular microenvironment perturbations and is mediated by a specific class of cysteine protease named caspases [7]. After extracellular or intracellular apoptotic stimuli, caspases activate all molecular pathways necessary to eliminate damaged cells [8]. Conversely, necrosis is the result of accidental events induced by different stimuli such as osmotic variations, arrest of the supply of nutrients, protein denaturation and it is characterized by a robust inflammatory and immunogenic response [5][9][5,9].
Referring only to morphological criteria, cell death was initially classified as (i) apoptosis, PCD type I; (ii) autophagy, PCD type II; and (iii) necrosis, lacking PCD type I and II features [2]. Following advancements in biochemical approaches, the Nomenclature Committee on Cell Death (NCCD) has updated RCDs guidelines, by the addition and description of other forms of programmed cell death, whose molecular mechanisms and biological functions must be better clarified [5][6][10][5,6,10]. This classification subdivided the RCD into 12 classes on the basis of molecular mechanisms, morphological features, and immunomodulatory profile: intrinsic apoptosis, extrinsic apoptosis, MPT-driven necrosis, necroptosis, ferroptosis, pyroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent cell death, autophagy-dependent cell death, and immunogenic cell death [11]. It is now known that there is a molecular interconnectivity between the different RCDs, although each one is characterized by a specific signal transduction cascade and morphological, biochemical, and immunogenic features as biomarkers [11].
Among them, necroptosis is the first form of programmed necrosis described with a prominent role in multiple physiological and pathological conditions [6]. It is induced by different extracellular or intracellular stimuli and detected by specific death receptors, PRRs and ZBP1 [10][11][12][10,11,12]. Numerous evidences suggest the involvement of necroptosis in the pathogenesis of diseases including organ injury, neurodegenerative diseases and viral infections [12][13][12,13]. Furthermore, necroptosis seems to control the homeostasis of T cells by eliminating the abnormal and excess ones in caspase-8 deficient T cells [14]. In addition to these physio-pathological functions, necroptosis shows a contradictory role in cancer that needs to be better investigated. Indeed, several studies suggest its involvement as a cancer suppressor, while others as cancer promoter [15][16][15,16]. The activation of markers involved in regulated necrosis pathways, such as ROS production, inhibition of tumor immunity, and activation of inflammation, leads to cancer progression through angiogenesis and metastasis. However, specific markers of necroptosis remain to be identified. Analysis of necroptosis, especially in vivo and in human tissue samples, is prevented by the lack of molecular markers [15][17][15,17].
2. Involvement of Necroptotic Mediators in Cancer
It is not surprising that RIPK1, RIPK3, and MLKL in cancer play a central role in modulating necroptosis. Functional mutations in the necroptotic machinery can compromise the death of cancer cells and influence the prognosis due to changes in interactions between RIPKs and other proteins [18]. Upregulation and downregulation of key necroptotic members have been found in many cancer types [16] (Table 1Table 2).
Table 12. Deregulated expression of necroptotic factors.
| Cancer Model | Dysregulated Expression | References | |
|---|---|---|---|
| AML | RIPK3 downregulation | [19] | [86] |
| Breast | RIPK3 and MLKL downregulation | [20] | [87] |
| Colorectal | RIPK3 downregulation | [21] | [88] |
| Cervical Squamous Cell Carcinoma |
MLKL downregulation | [22] | [89] |
| Gastric | MLKL downregulation | [23] | [90] |
| Glioblastoma | RIPK1 upregulation | [24] | [91] |
| Head and Neck Squamous Cell Carcinoma |
RIPK1 downregulation | [25] | [92] |
| Liver | RIPK1 downregulation | [26] | [93] |
| Lung | RIPK1 upregulation | [27] | [94] |
| Ovarian | MLKL downregulation | [28] | [95] |
| Melanoma | RIPK3 downregulation | [29] | [96] |
| Pancreatic adenocarcinoma early-stage | MLKL downregulation | [30] | [97] |
| Pancreatic ductal adenocarcinoma | RIPK1, RIPK3, and MLKL upregulation | [31] | [98] |
Over the years, interest in the correlation between gene mutations associated with RIPK1 and cancer has grown. Furthermore as reported by the COSMIC database about the somatic mutations in cancer, RIPK1 gene is not significantly mutated in a specific tissue, rather, the identified mutations are: about 36% missense substitutions, 10% synonymous substitutions, 5% nonsense substitutions, 0.4% frameshift insertions, 0.2% frameshift deletions, and 9% of mutations without detailed information available [32][99]. Ripk1 belongs to those genes with a part probably implicated in cancer and the ICGC database reported genomic and proteomic mutations in cancer distribution (https://dcc.icgc.org/). In general, RIPK1 maintains normal expression levels in many cancer types but in glioblastoma or lung cancer its expression has been found to be upregulated, affecting cancer prognosis negatively [16]. RIPK1 protein expression seems to be higher in glioblastoma grade IV, the most common adult malignant brain tumor, compared to lower grade glioma (I–III). RIPK1 overexpression with a consequent activation of NF-ĸB, upregulation of MDM2 and inhibition of p53 pathway lead to a worse prognosis, resistance to DNA damage and malignant phenotype [24][91]. According to the tumor-promoting role of RIPK1 in glioblastoma, a recent study suggests the oncogenic function of RIPK1 in the lung epithelial cells that have acquired genetic mutations and epigenetic modifications caused by carcinogens [27][94]. An increase in RIPK1 expression induced by cigarette smoke carcinogens suppresses ROS accumulation and MAPK-mediated cytotoxicity in DNA-damaged bronchial epithelial cells by facilitating malignant transformation [27][94]. RIPK1 is also a critical regulator of hepatocyte survival that cooperates with NF-ĸB to control TNFR1-dependent and -independent chronic liver inflammation and cancer [26][33][93,100]. Under physiological conditions, RIPK1 synergizes with NF-ĸB to prevent hepatocyte apoptosis, chronic liver disease and cancer. However, in the absence of NEMO, RIPK1 activity drives hepatocyte apoptosis in a kinase-dependent manner, resulting in HCC development [33][100]. Additionally, low expression of RIPK1 and TRAF2 in HCC patients undergoing liver resection or transplantation predicted poor prognosis [26][93].
The tumor stage can also influence RIPK1 expression levels, for example in HNSCC the metastatic stage shows reduced transcriptional and protein expression levels of RIPK1 compared to primary tumor and this could be associated to epigenetic events such as the methylation status in the RIPK1 promoter. Hypermethylation of a specific site reduces the binding of the transcription factor ARID3A to the promoter of Ripk1 resulting in a downregulation of the protein with compromised cell integrity [25][92].
Fusion genes have a significant evolution in the architecture of a gene and play a key role in tumorigenesis since these represent an important class of somatic alterations in cancer [34][101]. RIPK1 belongs to the genes involved in translocations which are found in adenocarcinoma of breast, prostate, and ovary (Table 2Table 3). RIPK1 has been found also in Astrocytoma, Grade III-IV/glioblastoma in which translocation t(6;6) (p25:p25) occurs. In order to suggest therapeutic approaches and predict kinases inhibitors it is interesting to unravel the molecular roles of these fusion transcripts involving protein kinase genes [34][101].
Table 23. RIPK1 fusion gene involved in tumorigenesis
| Morphology-Topography | Genes | Abnormality | References | |
|---|---|---|---|---|
| Breast Adenocarcinoma | RIPK1/BCKDHB | t(6;6) (p25;q14) | [34] | [101] |
| Breast Adenocarcinoma | RIPK1/NQO2 | t(6;6) (p25;p25) | [35] | [102] |
| Prostate Adenocarcinoma | FARS2/RIPK1 | t(6;6) (p25;p25) | [34][36] | [101,103] |
| Brain Astrocytoma, Grade III-IV/Glioblastoma | RIPK1/SERPINB9 | t(6;6) (p25;p25) | [37] | [104] |
| Ovary Adenocarcinoma | RIPK1/TTC27 | t(2;6) (p22;p25) | [34] | [101] |
Finally, RIPK1 also belongs to a set of 21 genes that are aberrant expressed and revealed to be more likely involved in the metastatic process of monosomy 3 in uveal melanomas [38][105].
In addition to RIPK1, the necroptotic mediators RIPK3 and MLKL are also involved in cancer and have influence on prognosis. It is already known that most cancer cells lose RIPK3 expression, so the downregulation or deletion of RIPK3 during tumorigenesis accompanied by necroptosis resistance determines a poor prognosis [16][39][16,106]. This deregulation could be mediated by oncogenes such as BRAF and AXL/TYRO3 able to control one of the transcription factors (Sp1) of RIPK3 and the promoter by methylation state [40][41][107,108]. A hypo-methylation of RIPK3 promoter that restores the normal expression of the kinase leads to an increase in sensitivity to chemotherapy by improving anticancer treatment [39][106]. Interestingly, in some colon cancer cell lines the mRNA expression levels of both RIPK1 and RIPK3 decrease due to hypoxia but not by promoter methylation status leading to a worse prognosis [42][109]. Additionally, RIPK3 is downregulated in breast, colorectal, melanoma, and AML cancers [16]. Although the mechanisms are still unclear, RIPK3 and MLKL deficient breast cancer cells are related to a reduced expression of the genes involved in interferon-α and interferon-γ responses [20][87]. RNA-Seq analysis using CD34+ bone marrow cells from patients with myelodysplastic syndromes or chronic myelomonocytic leukemia revealed overexpression of MLKL and its association with the severity of anemia. The increased level of RIPK1 expression supports its role as a mediator of inflammation, defining it as a predictor of worse overall survival although the mechanism remains unclear [43][110]. AML patients with CD34+ leukemia cells show reduced RIPK3 expression while the expression of RIPK1 is not affected leading to suppressed apoptosis, necroptosis, and NF-ĸB pathway [19][86]. Moreover, an additional study suggested that RIPK1/RIPK3 inhibition may be an effective treatment for AML patients when combined with specific chimeric antigen receptor T cells (that express high levels of IFN-γ) or other differentiation inducers in order to repress leukemogenic capacity of AML cells [44][111]. Tumor progression and reduced survival in patients with early-stage pancreatic adenocarcinoma, gastric, ovarian, and cervical squamous cancer is associated to low MLKL expression [22][23][89,90]. The downregulated MLKL expression could lead to inhibition of the necroptotic phenomenon and therefore determine a poor prognosis underlying its role as prognostic biomarker in cancer. In contrast RIPK1, RIPK3, and MLKL are widely expressed in human pancreatic ductal adenocarcinoma associated with robust manifestation of chemokines resulting in tumor promotion [31][98].
3. Conclusions
Cell death has a great impact in the regulation of physiological and pathological phenomena. The recent discovery of a new form of programmed necrosis has opened the way to develop pro-necroptotic drugs for anti-cancer therapy. More studies are needed to contextualize the role of necroptosis in different types of cancer. Since necroptotic phenomenon can take the dual function of tumor suppressor and tumor promoter, it could be interesting to manipulate its mediators to direct cancer cells towards death by improving patient survival. Using the members of the necroptotic system as biomarkers in cancer can be useful for prognostic information. Many in vitro studies have been conducted so far, so more efforts are needed to explore necroptotic mechanisms in vivo to achieve more efficient anticancer therapies.