Most animal cell types have the ability to undergo turnover at different rates throughout the organism's life span, dying either accidentally or in a deliberate manner. If a cell suffers from irreparable structural or organelle damage, it most likely passively disintegrates and dies. In this case, the plasma membrane ruptures and the noxious intracellular components are released into the extracellular matrix, where they trigger an inflammatory response. However, if a cell sustains non-fatal damage, or if it is too old, contains dysfunctional organelles, has suffered oxidative damage, etc, it is deliberately eliminated through an active, physiologically-regulated process of cell death termed regulated cell death (RCD), which is not accompanied by an inflammatory response. RCD plays beneficial physiological roles in development and in systems maintenance, but can become malignant and lead to pathological conditions when it is impaired, insufficient or in excess.
In the context of liver injury and disease, RCD is pivotal in directing the severity and outcome of the disease. Hepatocyte death is a critical event in the progression of disease due to resultant inflammation, which may lead to fibrosis, cirrhosis, and other morbidities if not treated in a timely manner.
- hepatocytes
- apoptosis
- necrosis
- necroptosis
- pyroptosis
- ferroptosis
1. Apoptosis
Apoptosis, derived from the Greek word for falling leaves, is the most comprehensively studied and described form of programmed cell death. Apoptosis can be triggered either intrinsically or extrinsically [1]. Both pathways lead to the activation of the executioner caspases 3 and 7 (CASP3, CASP7), resulting in proteolysis, nuclear fragmentation, and apoptotic cell death. Cellular injury such as DNA damage, starvation, or oxidative stress can activate the intrinsic apoptotic pathway [1]. The intrinsic pathway of apoptosis is defined by the nomenclature committee on cell death (NCCD) as a form of regulated cell death that is activated by perturbations in the intracellular and extracellular microenvironment characterized by mitochondrial outer membrane pore formation (MOMP) and precipitated by executioner caspases [1]. MOMP is controlled by the balance between the pro-apoptotic and anti-apoptotic members of the B cell lymphoma-2 (Bcl-2) family of regulator proteins [2]. In apoptosis, the activator Bcl-2 protein, BH3-interacting domain death agonist (BID), undergoes post-transcriptional modification and cleavage, forming tBID. Then, tBid translocates to the mitochondria and interacts with the mitochondrial pool of proapoptotic Bcl-2 members, BCL2-associated X apoptosis regulator (BAX), and/or BCL2 antagonist/killer (BAK) [1]. This results in conformational changes, leading to MOMP and mitochondrial release of apoptotic constituents such as cytochrome c and second mitochondria-derived activator of caspase/direct inhibitor of apoptosis binding protein with low pl (Smac)/DIABLO. Cytochrome c binds apoptotic peptidase activating factor 1 (APAF1) and pro-caspase 9 (pro-CASP9), forming the apoptosome [3]. The apoptosome advances the self-activation of CASP9, ultimately resulting in activation of executioner caspases (CASP3 and CASP7), causing cell death (Figure 1A) [1].
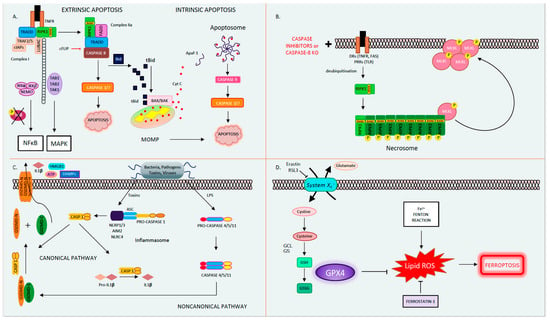
Figure 1. The four different types of regulated cell death. (A). Apoptosis. Apoptosis is a form of orderly programmed cell death, occurring both via an extrinsic pathway and an intrinsic pathway. In the extrinsic pathway, after a DR is engaged (TNFR as an example depicted here) by its ligand (here, TNF), it oligomerizes and binds the adaptor TRADD, the E3 ubiquitin ligases TRAF2/5, cIAPs, and LUBAC, as well as the kinase RIPK1 to form Complex I. Then, RIPK1 becomes polyubiquitinated and serves as a scaffolding protein for the further recruitment of TAK1-TAB1/2 and IKKα/β-NEMO. TAK1 activates the MAPK pathway, while phosphorylation and the subsequent degradation of IκB leads to NFκB activation and the transcription of pro-survival/inflammatory genes. If RIPK1 is deubiquitinated, it forms Complex IIa, consisting of FADD, TRADD, RIPK1, and caspase 8. Caspase 8 becomes activated in Complex IIa, in turn activating its downstream executioner caspases 3 and 7, leading to apoptosis. In the intrinsic pathway, caspase 8 mediates the cleavage of Bid into tBid. tBid translocates to the mitochondria where it binds BAX/BAK, leading to MOMP and Cytochrome C release. Cytochrome C binds Apaf-1, releasing its auto-inhibitory hold and allowing for its oligomerization, apoptosome formation, and caspase 9 activation. In turn, caspase 9 activates the executioner caspases 3 and 7, leading to apoptosis. (B). Necroptosis occurs when death receptors (such as TNF and FAS) or pattern recognition receptors (such as TLR2/3) are activated in the presence of pan-caspase inhibitors to block apoptosis. In this instance, deubiquitinated RIPK1 forms Complex IIb (also known as the necrosome) by associating with RIPK3 through their shared RIP homology interaction motif (RHIM) to form the necrosome. RIPK3 auto-phosphorylates, polymerizes, and in turn recruits and phospho-activates MLKL to the necrosome (pMLKL). Subsequently, pMLKL translocates to the plasma membrane where it forms a tetramer and permeabilizes the cell membrane causing oncolysis. (C). Pyroptosis. Pyroptosis occurs through two pathways. In the canonical pathway, upon the recognition of external toxins or pathogens, the supramolecular inflammasome (consisting of NLRPs, NLRC4, AIM2, the adaptor ASC, and pro-caspase 1) forms. Pro-caspase 1 undergoes autocatalytic cleavage to form activated caspase 1. In turn, caspase 1 cleaves pro-IL-1β into active IL-1β and GSDMD into GSDMD-C and GSDMD-N. GSDMD-N translocates to the plasma membrane where it forms a pore, releasing DAMPs (such as ATP and HMGB1) and IL-1β. In the noncanonical pathway, the detection of LPS directly activates caspases 4/5/11, which then cleaves GSDMD to form the membrane pore and induce pyroptosis. (D). Ferroptosis. Ferroptosis is a type of regulated cell death that is driven by intracellular lipid peroxidation and is highly dependent on ROS generation and iron availability. Cystine enters the cell by the cystine/glutamate antiporter (System Xc-). Within the cell, cystine is reduced to cysteine, and glutamate cysteine ligase (GCL) catalyzes the synthesis of γ-glutamyl cysteine from glutamate and cysteine. Glutathione synthase (GS) generates glutathione (GSH) by adding glycine. GSH reduces free radicals when it is reduced to GSSG by glutathione peroxidase-4 (GPX4). GPX4 and other free radical scavengers such a ferrostatin-1 inhibit lipid peroxidation (lipid-ROS). In the absence of GPX4, lipid peroxides accumulate driving ferroptosis. Abbreviations: AIM2: absent in melanoma-2, APAF1: apoptotic peptidase activating factor 1, ASC: apoptosis-associated speck-like protein containing a CARD, ATP: adenosine triphosphate, BAK: BCL2 antagonist/killer, BAX: BCL-2-like protein 4, CASP: caspase, cIAPs: cellular inhibitor of apoptosis, DAMP: danger-associated molecular patterns, DR: death receptor, FADD: Fas associated via death domain, GCL: glutamate cysteine ligase, GPX4: glutathione peroxidase-4, GS: glutathione synthase, GSDMD: gasdermin D, IL: interleukin, HMGB1: high mobility group box 1, IKKα/β: IKB kinase, LPS: lipopolysaccharide, LUBAC: linear ubiquitin chain assembly complex, MAPK: mitogen-activated protein kinase, MLKL: pseudokinase mixed lineage domain-like, MOMP: mitochondrial outer membrane pore formation, NEMO: NF kappa B essential modulator, NLRC: NLR family CARD domain-containing protein, NLRP: NOD-like receptor family, pyrin domain-containing, RHIM: RIP homology interaction motif, RIPK1/3: receptor-interacting protein kinase 1/3, ROS: reactive oxygen species, TAB1/2: TAK binding protein, TAK1: transforming growth factor-activated kinase-1, TLR2/3: Toll-like receptors 2/3, TNF: tumor necrosis factor, TNFR: tumor Necrosis Factor Receptor, TRADD: TNFR-associated death domain, TRAF2/5: TNF receptor-associated factors 2/5.
The extrinsic pathway of apoptosis is initiated by perturbations of the extracellular microenvironment and mostly driven by stimulation of transmembrane receptors such as death receptors (DRs) or pattern recognition receptors (PRRs) [1]. Ligation to DRs, such as Tumor Necrosis Factor Receptor (TNFR), FAS, tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL), or PRRs such as Toll-like receptors (TLRs), results in the assembly of a multiprotein complex called the Death-inducing signaling complex (DISC) or complex 1, which regulates caspase 8 (CASP8) activity and downstream interactions [1]. Multiple proteins form complex 1, including receptor-interacting protein kinase-1 (RIPK1), cellular inhibitor of apoptosis 1 and 2 (cIAP1 and 2), TNF receptor-associated factors 2 or 5 (TRAF2 or TRAF5), and the adaptor TNFR-associated death domain (TRADD). It is well known that DR ligation and in particular TNFR ligation does not always result in cell death. RIPK1 has a direct influence on the outcome of TNFR activation, resulting in pro-survival vs. pro-death pathways for the affected cell depending on its post-translational modifications [4][5]. The E3 ubiquitin ligases cellular inhibitor of apoptosis (cIAPs) catalyze the K63 polyubiquitination of RIPK1, forming a platform that facilitates the activation of the Nuclear factor-κΒ (NFκΒ) pathway through transforming growth factor-activated kinase-1 (TAK1) and TAK-1 binding proteins (TAB2 and 3) [6]. NFκΒ activation subsequently leads to the transcription of pro-survival genes that prevent cell death [6][7]. On the other hand, if RIPK1 is deubiquitinated, it is released from complex 1 to form complex 2, where it binds to FADD and CASP8 [6][7]. In lymphocytes and other Type I cells, CASP8 cleavage leads to apoptotic cell death by the subsequent activation of executioner CASP3 and CASP7 [8]. However, in type II cells such as hepatocytes, the formation of complex 2 is constitutively blocked by x-linked inhibitor of apoptosis (XIAP), NFκΒ target genes such as TNF alpha induced protein 3 (TNFAIP3, also known as A20), and Fas-associating protein with death domain-like interleukin-1 -converting enzyme (FLICE) inhibitory protein (c-FLIP) [1][9]. c-FLIP, a catalytically inactive close relative of CASP8, exists in a long form (cFLIPL) and short form (c-FLIPS) and is a regulator of CASP8 activation. cFLIPL heterodimer binds to CASP8 (CASP8-cFLIPL) promoting CASP8 oligomer assembly, while the short heterodimer cFLIPS (CASP8-cFLIPS) blocks CASP8 enzymatic activity [10]. It is important to note that cFLAR, the gene encoding cFLIP, is under the transcriptional control of NFκΒ (Figure 1A) [1]. In type II cells, extrinsic apoptosis requires the cleavage of BID to tBID and its translocation to mitochondria, resulting in MOMP-driven apoptosome formation and cell death as described above. Therefore, in hepatocytes, the amplification of the death signal and mitochondrial engagement is necessary for DR-induced cell death [9].
2. Mitochondrial Permeability Transition-Driven Necrosis
Necrosis is a form of cell death characterized by cell swelling and loss of plasma membrane integrity [1]. Mitochondrial Permeability Transition (MPT)-driven necrosis is a form of regulated cell death (RCD) initiated by toxins or perturbations in the cellular microenvironment due to oxidative stress, which results in the abrupt loss of mitochondrial membrane potential and cell membrane rupture [1]. Morphologically, necrosis contrasts with apoptosis, such that necrosis results in nuclear condensation, cell shrinkage, and blebbing, although secondary necrosis following apoptosis has been described [11]. Following necrosis, cellular contents, including cytotoxic and pro-inflammatory factors such as danger-associated molecular patterns (DAMPs), are released into the extracellular space [12]. The exact mechanisms leading to MPT have not been fully elucidated; however, this pore is proposed to be composed of dimers of the ATP synthase complex, which can be opened by interacting with the mitochondrial protein cyclophilin D (CypD) [13]. CypD inhibitors, such as cyclosporin A (CsA), or knockdown and knockout (KO) of CypD have shown promise in preventing necrosis in animal models of oxidative stress such as renal, cardiac, liver, and neurologic ischemia reperfusion, neurodegeneration, cancer, as well as acetaminophen (APAP) toxicity and hepatic steatosis [14][15][16]. While the role of CypD in MPT is experimentally evident, its translational relevance and application to human disease has been called into question due to negative results with trials of cyclosporin A (CsA) preventing cardiac myocyte death in humans undergoing cardiac catheterization [17]. Therefore, much remains unknown regarding the precise mechanism and signaling events leading to regulated necrosis from MPT.
3. Necroptosis
The best known and most extensively studied pathway of regulated necrosis is necroptosis, which was initially described when a shift in the cell death mode from apoptosis to necrosis was observed upon TNFR stimulation with TNF in the presence of caspase inhibitors [18]. Necroptosis is initiated by DRs, PRRs such as TLRs, or intracellular sensors such as Z-DNA binding protein 1 (ZBP1/DAI). This RCD pathway occurs with caspase inhibition and necessitates the proteins RIPK1, RIPK3, and the pseudokinase mixed lineage domain-like (MLKL) [19][20][21][22][23]. Complex 1 is created following DR ligand binding. In several cell types, when CASP8 is inhibited, deubiquitinated RIPK1 does not associate with complex 2 but alternatively binds to RIPK3 through the shared RIP homology interaction motif (RHIM) [22]. Then, the necrosome complex is formed, which is followed by the RIPK3 recruitment of MLKL. RIPK3 phospho-activates MLKL (forming pMLKL), driving its translocation to the plasma membrane, where it oligomerizes and induces pore opening and necroptotic cell death [22][24]. Necroptotic cell death is kept in check by CASP8-mediated cleavage of RIPK1 and RIPK3, such that in the presence of CASP8, the cell death mode defaults to apoptosis [25][26]. Accordingly, an inhibition of CASP8 is imperative for necroptosis to occur, raising the question regarding the contribution of necroptosis to the pathology of human disease conditions where caspases are intact and not inhibited (Figure 1B) [27]. The kinase activity of RIPK1 is necessary for necroptosis and RIPK1-dependent apoptosis to proceed but is nonessential for its pro-survival function [28]. The kinase activity of RIPK3, which activates MLKL, is similarly essential for necroptosis [23]. The role of the RIPK proteins, MLKL and necroptosis in liver disease and liver cell death has garnered much controversy in the past few years [27][29]. Complicating matters further are the intricacies and the various non-necroptotic functions of these proteins and in particular the role of RIPK1 and RIPK3 in apoptosis and inflammation [23][29]. Under basal conditions, RIPK3 is undetectable in liver cells [30], and its induction is controversial; therefore, the role of necroptosis in hepatocyte cell death in liver diseases is under extensive investigation [27]. However, RIPK3 has been detected in non-parenchymal cells (NPCs), and MLKL is expressed in all liver cell types. MLKL also has non-necroptosis functions; for example, it has been shown to inhibit autophagy and to play an important role in vesicle trafficking. [31][32]. Therefore, the possibility of necroptosis contributing as a mode cell death in these cell types in the liver remains an intriguing idea that warrants further investigation [29][33][34][35][36].
4. Autophagy and Autophagy-Dependent Cell Death
Autophagy is an intracellular waste degradation pathway that occurs through the formation of the autophagosome. Autophagy plays a pivotal role in cell survival [37]. Autophagy is activated in response to cellular stress, mediating protective rather than cytotoxic effects [1]. Defects in the autophagic machinery have been associated with various pathological settings. Blocking autophagy commonly expedites the stress response of cells during pathological conditions [1]. In fact, the concept of autophagic cell death is thought to be rather controversial. Components of the autophagy machinery closely interact with the apoptotic machinery, and in certain contexts and specific models, they have been associated with the promotion of cell death. Much of the discussion on autophagy-dependent cell death occurs during development and has been studied in Drosophila [1]. However, certain mammalian examples exist. For example, the deletion of autophagy-related gene 7 (Atg7) prevents neurotoxicity in mouse models of hypoxia–ischemia, or the blockade of Atg5 prevents the neurotoxicity of cocaine [1]. In liver disease models where autophagy plays a prominent role, such as alcoholic steatohepatitis, autophagy has mostly proven to be a protective rather than death-inducing mechanism. During autophagy, double membrane autophagosomes fuse with the lysosome to form autolysosomes within which autophagic cargo is degraded by hydrolases [1]. The KO of ATG genes expedites cell death in most models [38]. However, in particular instances, autophagy eventually leads to RCD [1]. The term "Autophagic cell death" is advised to be used when markers of autophagy and autophagic degradation substrates are elevated. Most importantly, when cell death can be prevented with autophagy inhibition [38]. The interaction of autophagy with apoptotic cell death, necroptosis, and ferroptosis has been extensively studied in various organs, including the liver [39][40][41].
5. Pyroptosis
Pyroptosis is a profoundly inflammatory mode of RCD related to the innate immune system [42]. It has evolved to remove intracellular pathogens and has a distinct morphology associated with cell bursting. The canonical pathway of pyroptosis occurs when inflammasome sensors, NOD-like receptor family, pyrin domain-containing-1 and 3 (NLRP1, NLRP3), or absent in melanoma-2 (AIM2) are stimulated by pathogens, pathogen-associated molecular patterns (PAMPs), and DAMPs and recruit CASP1 to activate Gasdermin D (GSDMD), which forms a pore in the plasma membrane [1]. In the non-canonical pathway, cytosolic LPS and PAMPs stimulate CASP4, 5, and 11 directly, which in turn cleave GSDMD. Then, activated GSDMD, the main conduit of pyroptosis, binds membrane phospholipids and initiates pore formation, resulting in cell death (Figure 1C) [43][44][45]. The contribution of pyroptosis to liver disease and in particular Non-Alcoholic Steatohepatitis (NASH) is the topic of intense research in Hepatology [46][47]. Unrepressed NLRP3 activation has been shown to result in shortened survival, severe liver inflammation, and hepatic stellate cell (HSC) activation, leading to collagen deposition and liver fibrosis [48].
6. Ferroptosis
Ferroptosis is another form of RCD characterized by glutathione (GSH) depletion and severe iron-dependent lipid peroxidation in a Fenton-like manner, resulting in reactive oxygen species (ROS) production and cell death [49][50]. Ferroptosis has been shown to contribute to cancer-related cell death, as well as ischemia reperfusion injury, neurologic diseases, and acute kidney injury [51]. This form of RCD occurs independently of caspases, necroptosis, autophagy, and pyroptosis. However, it does have subcellular elements reminiscent of necrosis and could be associated with a release of DAMPs [1]. The presence of miniature mitochondria with compressed mitochondrial densities with vanished cristae are morphological hallmarks of ferroptosis [52][53]. The GSH-dependent enzyme glutathione peroxidase-4 (GPX4) is the main endogenous inhibitor of ferroptosis due to its ability to limit lipid peroxidation by reducing lipid peroxides to alcohols [1][54]. The inhibition of GPX4 activity can lead to the accumulation of lipid peroxides, which is a marker of ferroptosis (Figure 1D). Many of the signaling events and effectors of ferroptosis have been elucidated by performing experiments using specific activators (such as RSL3 and erastin) and inhibitors (such as ferrostatins) of ferroptosis. For example, the commonly used agent, Erastin, activates ferroptosis by inhibiting System Xc−, the cystine/glutamate antiporter system, which then decreases intracellular cysteine and limits GSH synthesis [51]. The role of ferroptosis has been newly investigated in various models of liver disease and will be further discussed in our review.
7. Other Modes of Cell Death
Other forms of cell death such as NETosis, parthanatos, entotic cell death, and lysosome-dependent cell death have been described in the literature, which are not covered here due to the lack of robust data on these cell death subroutines in liver diseases [1].
References
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541, doi:10.1038/s41418-017-0012-4.
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80, doi:10.1038/cdd.2017.186.
- Riedl, S.J.; Salvesen, G.S. The Apoptosome: Signalling Platform of Cell Death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413, doi:10.1038/nrm2153.
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.L.; et al. Linear Ubiquitination Prevents Inflammation and Regulates Immune Signalling. Nature 2011, 471, 591–596, doi:10.1038/nature09816.
- O’Donnell, M.A.; Legarda-Addison, D.; Skountzos, P.; Yeh, W.C.; Ting, A.T. Ubiquitination of RIP1 Regulates an NF-ΚB-Independent Cell-Death Switch in TNF Signaling. Curr. Biol. 2007, 17, 418–424, doi:10.1016/j.cub.2007.01.027.
- Ashkenazi, A.; Salvesen, G. Regulated Cell Death: Signaling and Mechanisms. Annu. Rev. Cell Dev. Biol. 2014, 30, 337–356, doi:10.1146/annurev-cellbio-100913-013226.
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of Tumour Necrosis Factor Signalling: Live or Let Die. Nat. Rev. Immunol. 2015, 15, 362–374, doi:10.1038/nri3834.
- Sun, S.C. CYLD: A Tumor Suppressor Deubiquitinase Regulating NF-B Activation and Diverse Biological Processes. Cell Death Differ. 2010, 17, 25–34, doi:10.1038/cdd.2009.43.
- Iorga, A.; Dara, L.; Kaplowitz, N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int. J. Mol. Sci. 2017, 18, 1018, doi:10.3390/ijms18051018.
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.R.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-Operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849, doi:10.1016/j.molcel.2016.02.023.
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714, doi:10.1038/nrm2970.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus Accessory Aspects of Cell Death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73, doi:10.1038/cdd.2014.137.
- Giorgio, V.; Von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabó, I.; et al. Dimers of Mitochondrial ATP Synthase Form the Permeability Transition Pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892, doi:10.1073/pnas.1217823110.
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in Mitochondrial Pathophysiology. Biochim. Biophys. Acta Bioenerg. 2010, 1113–1118, doi:10.1016/j.bbabio.2009.12.006.
- Ramachandran, A.; Lebofsky, M.; Baines, C.P.; Lemasters, J.J.; Jaeschke, H. Cyclophilin D Deficiency Protects against Acetaminophen-Induced Oxidant Stress and Liver Injury. Free Radic. Res. 2011, 45, 156–164, doi:10.3109/10715762.2010.520319.
- Wang, X.; Du, H.; Shao, S.; Bo, T.; Yu, C.; Chen, W.; Zhao, L.; Li, Q.; Wang, L.; Liu, X.; et al. Cyclophilin D Deficiency Attenuates Mitochondrial Perturbation and Ameliorates Hepatic Steatosis. Hepatology 2018, 68, 62–77, doi:10.1002/hep.29788.
- Song, K.; Wang, S.; Qi, D. Effects of Cyclosporine on Reperfusion Injury in Patients: A Meta-Analysis of Randomized Controlled Trials. Oxid. Med. Cell. Longev. 2015, 2015, doi:10.1155/2015/287058.
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of Caspases Increases the Sensitivity of L929 Cells to Necrosis Mediated by Tumor Necrosis Factor. J. Exp. Med. 1998, 187, 1477–1485, doi:10.1084/jem.187.9.1477.
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat. Chem. Biol. 2008, 4, 313–321, doi:10.1038/nchembio.83.
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-Activity Relationship Study of Novel Necroptosis Inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 5039–5044, doi:10.1016/j.bmcl.2005.07.077.
- Murphy, J.M.; Vince, J.E. Post-Translational Control of RIPK3 and MLKL Mediated Necroptotic Cell Death. F1000Research 2015, doi:10.12688/f1000research.7046.1.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227, doi:10.1016/j.cell.2011.11.031.
- Newton, K.; Manning, G. Necroptosis and Inflammation. Annu. Rev. Biochem. 2016, 85, 743–763, doi:10.1146/annurev-biochem-060815-014830.
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453, doi:10.1016/j.immuni.2013.06.018.
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Kőműves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by Caspase-8 Is Crucial for Limiting Apoptosis and Necroptosis. Nature 2019, 574, 428–431, doi:10.1038/s41586-019-1548-x.
- O’Donnell, M.A.; Perez-Jimenez, E.; Oberst, A.; Ng, A.; Massoumi, R.; Xavier, R.; Green, D.R.; Ting, A.T. Caspase 8 Inhibits Programmed Necrosis by Processing CYLD. Nat. Cell Biol. 2011, 13, 1437–1442, doi:10.1038/ncb2362.
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Questions and Controversies: The Role of Necroptosis in Liver Disease. Cell Death Discov. 2016, doi:10.1038/cddiscovery.2016.89.
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the Treatment of Human Diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722, doi:10.1073/pnas.1901179116.
- Dara, L. The Receptor Interacting Protein Kinases in the Liver. Semin. Liver Dis. 2018, 38, 73–86, doi:10.1055/s-0038-1629924.
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor Interacting Protein Kinase 1 Mediates Murine Acetaminophen Toxicity Independent of the Necrosome and Not through Necroptosis. Hepatology 2015, 62, 1847–1857, doi:10.1002/hep.27939.
- Yoon, S.; Kovalenko, A.; Bogdanov, K.; Wallach, D. MLKL, the Protein That Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017, 47, 51–65.e7, doi:10.1016/j.immuni.2017.06.001.
- Frank, D.; Vaux, D.L.; Murphy, J.M.; Vince, J.E.; Lindqvist, L.M. Activated MLKL Attenuates Autophagy Following Its Translocation to Intracellular Membranes. J. Cell Sci. 2019, 132, doi:10.1242/jcs.220996.
- Kaplowitz, N.; Win, S.; Than, T.A.; Liu, Z.X.; Dara, L. Targeting Signal Transduction Pathways which Regulate Necrosis in Acetaminophen Hepatotoxicity. J. Hepatol. 2015, 5–7, doi:10.1016/j.jhep.2015.02.050.
- Kasof, G.M.; Prosser, J.C.; Liu, D.; Lorenzi, M.V.; Gomes, B.C. The RIP-like Kinase, RIP3, Induces Apoptosis and NF-ΚB Nuclear Translocation and Localizes to Mitochondria. FEBS Lett. 2000, 473, 285–291, doi:10.1016/S0014-5793(00)01473-3.
- Xiaoqing, S.; James, L.; Navas, T.; Baldwin, D.T.; Stewart, T.A.; Dixit, V.M. RIP3, a Novel Apoptosis-Inducing Kinase. J. Biol. Chem. 1999, 274, 16871–16875, doi:10.1074/jbc.274.24.16871.
- Günther, C.; He, G.W.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Schleicher, U.; et al. The Pseudokinase MLKL Mediates Programmed Hepatocellular Necrosis Independently of RIPK3 during Hepatitis. J. Clin. Investig. 2016, 126, 4346–4360, doi:10.1172/JCI87545.
- Xie, Z.; Klionsky, D.J. Autophagosome Formation: Core Machinery and Adaptations. Nat. Cell Biol. 2007, 1102–1109, doi:10.1038/ncb1007-1102.
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 107–120, doi:10.1038/cdd.2011.96.
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 81–94, doi:10.1038/nrm3735.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 1425–1428, doi:10.1080/15548627.2016.1187366.
- Yin, X.M. Autophagy in Liver Diseases: A Matter of What to Remove and Whether to Keep. Liver Res. 2018, 109–111, doi:10.1016/j.livres.2018.09.001.
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen Blockade of TAK1 Triggers Caspase-8–Dependent Cleavage of Gasdermin D and Cell Death. Science 2018, 362, 1064–1069, doi:10.1126/science.aau2818.
- Man, S.M.; Kanneganti, T.D. Regulation of Inflammasome Activation. Immunol. Rev. 2015, 6–21, doi:10.1111/imr.12296.
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-Mediated Pyroptotic and Apoptotic Cell Death, and Defense against Infection. Curr. Opin. Microbiol. 2013, 319–326, doi:10.1016/j.mib.2013.04.004.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328, doi:10.3390/ijms20133328.
- Beier, J.I.; Banales, J.M. Pyroptosis: An Inflammatory Link between NAFLD and NASH with Potential Therapeutic Implications. J. Hepatol. 2018, 643–645, doi:10.1016/j.jhep.2018.01.017.
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D Plays a Key Role as a Pyroptosis Executor of Non-Alcoholic Steatohepatitis in Humans and Mice. J. Hepatol. 2018, 68, 773–782, doi:10.1016/j.jhep.2017.11.040.
- Wree, A.; McGeough, M.D.; Inzaugarat, M.E.; Eguchi, A.; Schuster, S.; Johnson, C.D.; Peña, C.A.; Geisler, L.J.; Papouchado, B.G.; Hoffman, H.M.; et al. NLRP3 Inflammasome Driven Liver Injury and Fibrosis: Roles of IL-17 and TNF in Mice. Hepatology 2018, 67, 736–749, doi:10.1002/hep.29523.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 165–176, doi:10.1016/j.tcb.2015.10.014.
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The Emerging Role of Ferroptosis in Non-Cancer Liver Diseases: Hype or Increasing Hope? Cell Death Dis. 2020, doi:10.1038/s41419-020-2732-5.
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, doi:10.1038/s41419-020-2298-2.
- Doll, S.; Conrad, M. Iron and Ferroptosis: A Still Ill-Defined Liaison. IUBMB Life 2017, 69, 423–434, doi:10.1002/iub.1616.
- Capelletti, M.M.; Manceau, H.; Puy, H.; Peoc’h, K. Ferroptosis in Liver Diseases: An Overview. Int. J. Mol. Sci. 2020, 21, 4908, doi:10.3390/ijms21144908.
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331, doi:10.1016/j.cell.2013.12.010.