Spinal muscular atrophy (SMA) is a congenital neuromuscular disorder characterized by motor neuron loss, resulting in progressive weakness. SMA is notable in the health care community because it accounts for the most common cause of infant death resulting from a genetic defect. SMA is caused by low levels of the survival motor neuron protein (SMN) resulting from SMN1 gene mutations or deletions. However, patients always harbor various copies of SMN2, an almost identical but functionally deficient copy of the gene. A genotype–phenotype correlation suggests that SMN2 is a potent disease modifier for SMA, which also represents the primary target for potential therapies. Increasing comprehension of SMA pathophysiology, including the characterization of SMN1 and SMN2 genes and SMN protein functions, has led to the development of multiple therapeutic approaches. Until the end of 2016, no cure was available for SMA, and management consisted of supportive measures. Two breakthrough SMN-targeted treatments, either using antisense oligonucleotides (ASOs) or virus-mediated gene therapy, have recently been approved. These two novel therapeutics have a common objective: to increase the production of SMN protein in MNs and thereby improve motor function and survival. However, neither therapy currently provides a complete cure. Treating patients with SMA brings new responsibilities and unique dilemmas. As SMA is such a devastating disease, it is reasonable to assume that a unique therapeutic solution may not be sufficient. Current approaches under clinical investigation differ in administration routes, frequency of dosing, intrathecal versus systemic delivery, and mechanisms of action. Besides, emerging clinical trials evaluating the efficacy of either SMN-dependent or SMN-independent approaches are ongoing. This review aims to address the different knowledge gaps between genotype, phenotypes, and potential therapeutics.
- spinal muscular atrophy
- survival motor neuron protein
- novel therapy
- clinical care
1. Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease characterized by devastating muscular wasting caused by the progressive degeneration of spinal motor neurons (MNs). Although recognized as a rare disease with an estimated global incidence of ~1/10,000 live births, SMA has been regarded as a noteworthy health issue because it is not only the second most common autosomal recessive inherited disorder, but is also the most common monogenic disease leading to infant mortality[1][2] [1,2]. The carrier frequency of SMA varies from 1 in 38 to 1 in 72, depending on the racial group, with the pan-ethnic average being 1 in 54[3] [3].
Figure 1 illustrates the genetic pathomechanism of SMA. All patients with SMA have an insufficient amount of a survival motor neuron (SMN) protein, which is encoded by two highly homogenous genes, survival of motor neuron 1 (SMN1) and its copy gene, SMN2. SMN2 is differentiated from SMN1 by one single nucleotide variant (CàT) in exon 7. This critical difference results in the preferential exclusion of exon 7 from most (~90%) SMN2 transcripts, termed SMN△7, which translates into truncated and unstable SMN protein. As a consequence, SMN2 can only generate ~10% of full-length (FL) SMN mRNAs and their product-functional SMN proteins (Figure 1A). While these FL-SMN2 transcripts can partially compensate for the loss of SMN1, it is reasoned that retained SMN2 copy numbers of patients determine the phenotypic severity (Figure 1B). However, such a phenotype–genotype correlation is not absolute, as recent studies have indicated that additional cellular mechanisms (e.g., positive or negative disease modifiers) might also involve the modulation of SMA clinical severity. For example, rare SMN2 variants (c.859G > C), as well as independent modifiers such as plastin 3 or neurocalcin delta, can further influence the disease severity[4][5][6] [4–6]. In brief, the loss of the SMN1 gene leads to SMA, whose severity is partially modified by various copies of SMN2.
Understanding SMN protein functions and mechanisms of action in subcellular contexts may elucidate potential pathways for therapeutic intervention. SMN is a multifunctional protein that is ubiquitously expressed in most somatic cells[7] [7]. The most appreciated canonical role of SMN is to serve as an essential component of small nuclear ribonucleoproteins (snRNPs) that form spliceosomes to process the pre-mRNA splicing[8][9] [8,9]. Studies on SMA animal models have revealed a direct correlation between the ability to assemble snRNPs and SMA phenotypes[10] [10]. SMN is also involved in DNA repair and mRNA transportation along MN axons[11] [11]. However, the multifaceted roles of SMN protein are still under investigation, and it is unclear how a deficiency in ubiquitously expressed SMN can selectively cause the dramatic MN degeneration. The cell autonomous effects related to deficient SMN are responsible for the MNs’ degeneration. However, this does not account for the full SMA phenotype, implicating not only the dysfunction of neural networks but other non-neuronal cell types involved in the disease process[12][13] [12,13]. For example, recent studies indicate that the MN survival and functionality of SMA animal and cellular models is highly dependent on glial cells, which play an important role in neuronal communication and neuroinflammation[14][15] [14,15]. These findings imply that SMA could also be a neuroinflammatory disease.
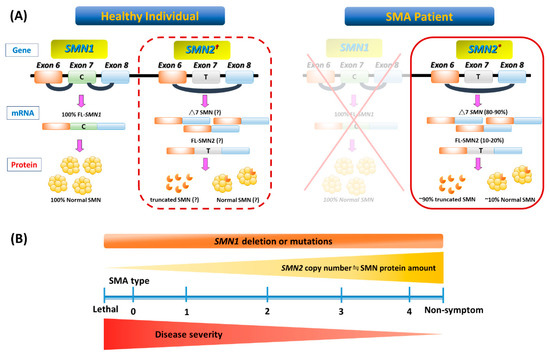
Figure 1. Genetic basis and phenotype-genotype correlation of spinal muscular atrophy (SMA). (A) In a healthy individual, full-length (FL) survival motor neuron (SMN) mRNA and protein arise from the SMN1 gene. Patients with SMA have homozygous deletion or mutation of SMN1 but retain at least one SMN2 (indicated with an asterisk in the solid-border box on the right). However, SMN2 can be dispensable in a healthy individual (indicated with an obelisk in the dotted-border box on the left). This single-nucleotide change in exon 7 (C-to-T) of SMN2 causes alternative splicing during transcription, resulting in most SMN2 mRNA lacking exon 7 (△7 SMN). About 90% of △7 SMN transcripts produce unstable truncated SMN protein, but a minority include exon 7 and code for FL, which maintains a degree of MN survival. (B) A continuous spectrum of phenotypes in SMA. Even with genetic confirmation of SMN1 absence or mutations in all patients, SMA presentation ranges from very compromised neonates (type 0) to adults with minimal manifestations (type 4) depending on the SMN2 numbers and FL SMN produced by each patient and modulated by potential disease modifiers that influence the final phenotype.
2. Clinical Characteristics of SMA
2.1. SMA Phenotypes and Classifications
SMA is classified into three main phenotypes based on age at the onset of symptoms/signs, and the highest motor function achieved[3][6][16] [3,6,16]. However, some patients with SMA are outliers on either end of the phenotypic spectrum. Subclassification has also been proposed in SMA types 1 and 3, and sometimes in the type 2 phenotype (Table 1 and Figure 1B). It should be kept in mind that regardless of the type of SMA, severity may vary and change over time, because the disease is a continuum.
Table 1. Classification and subtypes of spinal muscular atrophy.
|
SMA Type (Historical Name) |
OMIM |
Onset Age |
Motor Milestones Achieved |
Subclassification |
Natural History |
Other Features |
Estimated SMN2 Copies |
Estimated SMA Proportion |
|
Type 0 |
- |
Prenatal or at birth |
Never sits, never head control |
- |
Death < 1 mo if untreated |
Joint contractures, cardiac defect, facial diplegia, immediate respiratory failure after birth |
1 SMN2 copy in ~100% of patients |
Unclear, Maybe < 1% |
|
Type 1 (Werdnig-Hoffmann disease) |
253300 |
0–6 mo |
Never sits, some achieve head control |
1A: Onset < 1 mo, usually by 2 wk; head control absent 1B: Onset 1–3 mo; poor or absent head control 1C: Onset 3–6 mo; head control achieved |
1A: Death < 6 mo if untreated 1B and 1C: death < 2 yr if untreated |
1A: Very similar to type 0 SMA 1B and 1C: Tongue fasciculation, swallowing difficulties, early respiratory failure |
1 or 2 SMN2 Copies in ~80% of patients |
~60% |
|
Type 2 (Dubowitz disease)
|
253550 |
7–18 mo |
Sits but never stands |
2A: Sits independently, may lose the ability to sit in later life 2B: Sits independently, maintains the ability to sit According to functional level, decimal classification ranging from 2.1 to 2.9 |
Usually survive >2 yr; |
Proximal weakness, postural hand tremor, normal intellectual ability, kyphoscoliosis |
3 SMN2 copies in >70% patients |
~27% |
|
Type 3 (Kugelberg–Welander disease) |
253400 |
>18 mo |
Stands and walks |
3A: Onset between 18 and 36 mo 3B: Onset >3 yr |
Survival into adulthood |
May have hand tremor, resembles muscular dystrophy 3A: Scoliosis, usually early loss of ambulation |
3 or 4 SMN2 copies in ~95% of patients |
~12% |
|
Type 4 |
271150 |
10–30 yr, usually >21 yr |
Stands and walks |
- |
Survival into adulthood |
Usually preserved walking ability |
4 or more SMN2 copies in >90% |
~1% |
SMA: spinal muscular atrophy; mo: months; yr: years.
At one end of the spectrum, patients with type 0 SMA (categorized as type 1A by some authors) are usually associated with prenatal onset of signs such as a history of decreased fetal movements[17] [17]. These rare cases usually present with arthrogryposis multiplex congenita and have profound hypotonia and respiratory distress soon after birth. Life expectancy is hugely reduced, and if untreated, most cases are unable to survive beyond one month of age[1][18] [1,18].
Type 1 SMA patients account for more than 50% of the incidence of SMA. These patients are called non-sitters, who never achieve major motor developmental milestones such as sitting, standing, and walking in their lifetime. Notably, congenital heart defect is also a feature of severe SMA phenotype, especially in SMA types 0 and 1 [19]. If left untreated, most cases of mortality within the first two years of life are attributable to respiratory muscle dysfunction, with a median survival of 13.5 months[20] [20].
Patients with type 2 SMA (Dubowitz’s disease) are sitters who initially present with a delay in reaching developmental milestones for gross motor skills. Although these patients can maintain a sitting position unaided (“sitters”) and a few can even stand with leg braces, none can walk independently. Kyphoscoliosis usually develops, complicated with restrictive lung disease if there are no orthopedic interventions. Cough and ability to clear airway secretions are usually progressively compromised. A majority of patients with type 2 SMA can survive into adulthood. However, after entering their adolescence, these patients usually require aggressive supportive management, especially regarding gastrointestinal and respiratory complications[21][22] [21,22].
Patients with type 3 SMA (Kugelberg–Welander disease) are walkers who are able to stand unsupported and walk independently during their early life. These patients usually show profound symptom heterogeneity, so they are often misdiagnosed with myopathy or muscular dystrophy. The distribution of weakness is similar to that seen in patients with types 1 and 2 SMA, but the progression of weakness is much slower; some patients may eventually become wheelchair dependent at middle age[23] [23]. At the other end of the spectrum is the mildest adult-onset form, known as type 4 SMA, where individuals present onset symptoms—usually a weakness of lower extremities—after the second decade. Type 4 patients have a good prognosis with ambulation into adulthood and a mostly average life span[24] [24].
2.2. The Implication of Phenotypic Classification in SMA Clinical Trials
Efforts to better understand the natural history and define outcome measures pave the way for the readiness of SMA trials[25][26] [25,26]. Initially, the phenotype–genotype correlation encouraged the application of SMN2 copies as a criterion for patient enrollment in clinical trials[27] [27]. However, discordant cases certainly exist, and the prediction of phenotype solely through SMN2 copies is not always accurate in individual cases[6] [6]. Except for some disease modifiers, several prognostic factors have been identified[18] [18]. Although the severity-based classification has clinical advantages, it is not always adequate to provide prognostic information or to facilitate the stratification of SMA patients. Rather than the SMA phenotype, the ambulant status may be more relevant to the trajectory of disease progression[28] [28]. This approach acknowledges the SMA phenotypes as a continuum and focuses on the current functional status and the response to therapy. Furthermore, pulmonary function assessment may better reflect disease state than muscle strength[29] [29]. Nevertheless, repeat evaluations are imperative before assigning a patient to a specific SMA type. Particularly in patients with SMA types 2 and 3, the onset, time course, and extent of MN loss has not been well established, yet are vital in determining whether there is a specific therapeutic window for these patients with milder phenotypes.