Altered activity of fatty acid amide hydrolase (FAAH), an enzyme of the endocannabinoid system, has been implicated in several neuropsychiatric disorders, including major depressive disorder (MDD). It is speculated that increased brain FAAH expression is correlated with increased depressive symptoms.
- endocannabinoids
- fatty acid amide hydrolase
- major depressive disorder
- animal models of depression
1. Introduction
Major depressive disorder (MDD) is one of the leading causes of disability worldwide and one of the most prevalent psychiatric disorders, placing a large burden on society and healthcare costs [1]. The likelihood of relapse increases with each subsequent major depressive episode (MDE) [2]. Clinically, an MDE is characterized by symptoms, such as depressed mood, anhedonia, feelings of guilt or worthlessness, disruptions in cognitive function and self-harm or suicide [3]. It is widely acknowledged that MDD is a heterogeneous condition, and approximately 50% of individuals with MDD do not respond to first-line treatments, which primarily target monoamines [4][5][4,5]. This observation has turned the focus from the traditional monoamine hypothesis of depression to other neurochemical systems that may not be targeted by first-line, monoamine-elevating antidepressants, such as selective serotonin reuptake inhibitors (SSRIs) [6].
There is growing interest in the endocannabinoid system (ECS) as an alternative signaling pathway involved in MDD, since components of the ECS are highly expressed in fronto-limbic brain regions known to be affected in MDD, including the prefrontal cortex (PFC), hippocampus and amygdala [7]. Dysfunction in these regions has important functional implications in MDD. For example, dysfunction of the PFC affects many cognitive functions, such as decision making, attention, motivation and emotion regulation [8][9][8,9]. In the hippocampus, dysfunction may result in learning and memory impairments as well as compromised emotional and stress reactivity [9]. The amygdala is also known to be important for affective modulation and memory encoding [9]. Disruption of these key brain regions may result in functional impairment and emotional dysregulation in MDD. The ECS signaling pathway is composed of two main G protein-coupled receptors, cannabinoid 1 (CB1) and cannabinoid 2 (CB2) receptors, and two main endogenous cannabinoids (or endocannabinoids), N-arachidonylethanolamide (anandamide or AEA) and 2-arachidonylglycerol (2-AG), although other endocannabinoids are known to exist but are not as well-studied [10]. AEA and 2-AG are produced in the postsynaptic neuron on demand; that is, the endocannabinoids are not stored in vesicles, but rather they are biosynthesized and released as required [10]. AEA is synthesized from N-arachidonoyl-phosphatidyl-ethanolamine (NAPE) by N-acylphosphatidylethanolamine-selective phospholipase D (NAPE-PLD), whereas 2-AG is synthesized from sn-1-Acyl-2-arachidonylglycerol by diacylglycerol lipase (DAGL). NAPE-PLD and DAGL are stimulated by high calcium concentrations; therefore, calcium influx into the postsynaptic neuron is thought to trigger biosynthesis of AEA and 2-AG [10]. The endocannabinoids are then released into the synaptic cleft and bind, in a retrograde fashion, to CB1 and CB2 receptors that are located on axon terminals on the presynaptic neuron (Figure 1) [7]. The mechanism by which endocannabinoids are released into and taken up from the synaptic cleft, whether it is by passive diffusion across the plasma membrane or whether there is a membrane transporter involved, or both, is a matter of current debate (for a review of endocannabinoid transport, see Fowler [11]).
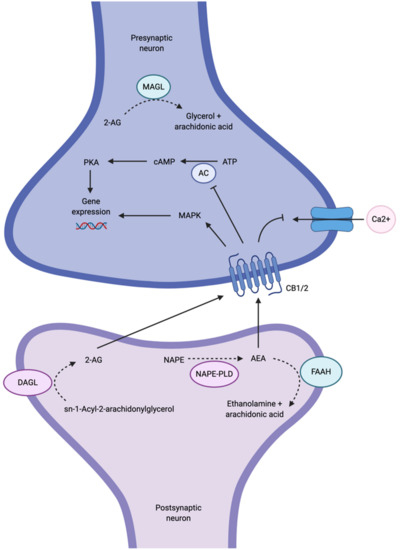
Although AEA and 2-AG are ligands for both CB1 and CB2 receptors, CB1 receptors are primarily targeted by AEA as well as Δ9-tetrahydrocannabinol (Δ9-THC), the psychoactive ingredient in marijuana [5][12][5,12]. Conversely, CB2 receptors are primarily targeted by 2-AG. Both CB1 and CB2 receptors are coupled to Gi/o proteins that, when stimulated, inhibit adenylyl cyclase (AC) activity, subsequently inhibiting cyclic AMP (cAMP) and the protein kinase A (PKA) phosphorylation pathways, or activating mitogen-activated protein kinase (MAPK) (Figure 1) [10]. Both events have the ability to regulate gene expression [10]. The activation of Gi/o proteins also reduces internal calcium currents; they are thus capable of regulating neurotransmitter release [4]. The net effect of ECS activity on synaptic transmission is dependent on whether CB1 receptors are found on GABAergic (inhibitory) or glutamatergic (excitatory) terminals [4]. To terminate endocannabinoid signaling, AEA is metabolized by fatty acid amide hydrolase (FAAH) in the postsynaptic neuron, and 2-AG is metabolized by monoacylglycerol lipase (MAGL) in the presynaptic neuron [7].
FAAH metabolizes AEA into ethanolamine and arachidonic acid, thereby terminating endocannabinoid signaling [13]. FAAH possesses both amidase and esterase activity, which allows the enzyme to hydrolyze N-acylethanolamines (e.g., oleamide) and monoacylglycerols (e.g., 2-AG), respectively [13]. It was first reported in 1993 that FAAH additionally hydrolyzes AEA [14]. FAAH belongs to a family of enzymes, named amidases, that hydrolyze fatty acid amide bonds [13]. FAAH is an integral membrane serine hydrolase composed of two subunits, characterized by a core structure of a twisted β-sheet surrounded by 24 α-helices [15]. FAAH possesses five key regions that are important for its enzymatic function: (1) the membrane-binding cap, which anchors the enzyme into the cytoplasmic leaflet of the lipid bilayer; (2) the membrane-access channel through which lipophilic substrates access the active site from the membrane; (3) the substrate-binding pocket, where the acyl chain of the substrate resides during catalysis; (4) the active site, which contains the highly conserved Ser241-Ser217-Lys142 catalytic triad; and (5) the cytosolic access channel, which serves as an exit path for the polar head group of the substrate after hydrolysis [16]. Covalent inhibitors of FAAH bind at Ser241, which blocks the catalytic function of FAAH [17].
Mouse and human FAAH genes are located on chromosomes 4 and 1, respectively, and share 84% amino acid sequence homology [18]. A single-nucleotide polymorphism exists in the FAAH gene (rs324420, C385A), whereby threonine replaces proline at position 129. This mutation makes the protein more susceptible to proteolysis, resulting in reduced activity [19]. The wild-type genotype, C/C, is associated with higher FAAH expression and protein activity, whereas the variant genotype, C/A or A/A, is associated with lower FAAH expression and protein activity [20][21][20,21]. For example, individuals possessing the A/A genotype exhibit reduced stress reactivity [22][23][22,23]. Recent studies have suggested that the ECS undergoes epigenetic modulation by diet, stress, drugs, alcohol, smoking or exercise [24]. For example, reduced DNA methylation at the FAAH gene was found in a study of late-onset Alzheimer’s disease, resulting in increased FAAH protein activity [25]. Interestingly, patients with the most severe cognitive impairment were found to have lowest levels of methylation. The FAAH gene promotor is also known to possess an estrogen-responsive element resulting in estradiol-dependent transcriptional activation (via demethylation) of FAAH [18]. Moreover, many studies have shown bidirectional interactions between the endocannabinoid and monoamine systems. For instance, FAAH gene knockout in mice has been found to increase serotonergic tone 4-fold in the frontal cortex [26]. In turn, Best and Regehr showed that activation of 5-HT2-type receptors by serotonin is able to evoke endocannabinoid release [27]. CB1 stimulation has also been shown to increase the firing activity of dopaminergic neurons and increase synaptic dopamine concentration [28]. Increased dopamine D3 receptor mRNA was observed in the brains of FAAH knock-in C385A mice; that is, lower FAAH function was associated with a greater number of dopamine D3 receptors in the brain [29]. These findings are relevant insofar as they demonstrate the functional overlap of the endocannabinoid and monoamine systems, which is an important consideration for developing new pharmacotherapeutics for MDD.
Genetic deletion of FAAH in mice results in 15-fold higher brain levels of AEA [30]. FAAH is widely distributed in the central nervous system (CNS), most prominently in the cerebral cortex, hippocampal formation, amygdala and cerebellum, and its regional distribution is correlated with the distribution of CB1 receptors as well as with the highest rates of AEA degradation [13][31][32][33][13,31,32,33]. CB1 receptors are also widely distributed in the CNS (particularly within the cerebral cortex, hippocampus, amygdala, striatum and substantia nigra) and have functional roles in mood, anxiety, pain, motor regulation, appetite control and memory processing [5][34][35][5,34,35]. Importantly, the ECS possesses plasticity; that is, the ECS is modulated in response to both acute and chronic stimuli. For instance, chronic exposure to exogenous agonists such as Δ9-THC results in reduced CB1 receptor density and signaling [36].
Alterations of the ECS are implicated in several neuropsychiatric disorders, such as Parkinson’s disease, Alzheimer’s disease, anxiety and MDD [37]. Human and animal data have shown that the ECS is involved in emotion regulation and cognition [38][39][38,39]. In response to stressors, the breakdown of AEA by FAAH is initiated, reducing AEA-CB1 signaling [34]. Moreover, corticotropin-releasing hormone, a stress-responsive neuropeptide, increases FAAH activity and thereby reduces AEA concentrations [40]. Studies linking stress and ECS dysregulation are of particular interest, since it is known that exposure to stressors can increase the risk of psychological disorders, including MDD [41]. Given the role of ECS signaling in mood, anxiety and cognition, it is likely that the ECS is hypofunctional (e.g., reduced AEA-CB1 signaling) in MDD and that increasing ECS signaling could elicit antidepressant-like effects. For example, the CB1 receptor antagonist, rimonabant, has been associated with an increased risk of developing or exacerbating depressive symptoms in humans, and genetic deletion of CB1 receptors in mice elicits a depressive-like phenotype [42][43][42,43]. These findings have triggered an interest in ECS-altering drugs to restore AEA-CB1 signaling. One such class of agents are FAAH inhibitors, which work by inhibiting FAAH activity, consequently increasing AEA concentration and stimulating downstream CB1 receptor signaling.
FAAH inhibitors are of particular interest, because they lack the same side effects that exogenous CB1 receptor agonists elicit, such as hypomotility, hypothermia and catalepsy [44]. Moreover, FAAH inhibition does not display reinforcing effects [44]. However, that is not to say that FAAH inhibitors do not carry their own risk of side effects, such as presyncope, diarrhea, and headaches [45]. Several irreversible FAAH inhibitors have been assessed in patients with MDD in clinical trials and, in fact, some were terminated due to adverse events. During a Phase I clinical trial of BIA 10-2474 conducted by Bial Pharmaceuticals, one participant died and five participants were hospitalized in January 2016 due to adverse effects of BIA 10-2474 [46]. It was speculated that BIA 10-2474 resulted in off-target effects that led to neurotoxicity [47].
2. Discussion
First, of the studies assaying FAAH gene expression and protein levels, the majority (four out of six studies) of the literature showed increased FAAH gene expression and protein levels in the brain, particularly in the PFC, hippocamps, and striatum, when the animal manifested depressive symptomology. Furthermore, increased FAAH gene expression and protein levels are accompanied by increased depressive-like behavior as measured by tests such as the FST. As Huang et al. demonstrated, once FAAH is degraded, depressive-like behaviors are also attenuated [48][54]. Therefore, a positive relationship could be established between brain FAAH gene expression and protein levels and depressive-like behaviors. Greater FAAH gene expression and protein activity results in greater degradation of AEA and thereby reduced AEA-CB1 signaling. This relationship is consistent with studies of other ECS components. For example, it has been found that there is reduced AEA concentrations and increased CB1 receptors in rats displaying depressive symptomology [49][63]. It has also been observed that activation of CB1 receptors with exogenous agonists elicits antidepressant-like effects [50][51][77,78]. Therefore, it appears that normal AEA-CB1 signaling is important for healthy mood, and it can be indirectly regulated via FAAH. Interestingly, in contrast to the other studies, one study by Kirkedal and colleagues found the opposite relationship, where decreased FAAH mRNA and protein levels were observed in Flinders Sensitive Line rats [52][55]. A possible reason for the discrepant finding is that the authors took a hemispheric approach to analyzing ECS components, whereas other groups may have pooled data from both hemispheres. Pooling of data from both hemispheres would mask any differences in ECS components between each hemisphere. The discrepant finding could also be due to differences in the animal model and species strain used across studies; this study used Flinders Sensitive Line rats, a genetic model of depression, whereas another study assessing FAAH protein levels used Sprague–Dawley rats and a CMS model. Additionally, Hill et al. did not find any association between depressive-like state and FAAH enzymatic activity, but rather found that the TCA, imipramine, increased FAAH enzymatic activity only in the presence of CUS [7].
Second, 14 studies demonstrated that genetic knockout or pharmacological inhibition of FAAH elicited antidepressant-like effects, reduced depressive-like behavior, and restored normal behavioral phenotypes. An important distinction to make between genetic knockout of FAAH and pharmacological inhibition of FAAH is that, since knockout animals never produce the FAAH protein, the complete absence of FAAH may be different from animals that had normal FAAH function but were then treated with an inhibitor; thus, phenotypic outcomes may also be different. The data from the genetic knockout studies of FAAH are scarce and mixed. Of the two available genetic knockout studies, Naidu et al.’s investigation failed to find any initial significant effects of FAAH knockout or inhibition on depressive-like behavior, which is inconsistent with the results of the other studies [53][58]. However, once methodological changes were instituted (namely, increasing sample size and altering lighting conditions to resemble Gobbi et al.’s study [54][60]), Naidu and colleagues were able to find a significant reduction in depressive-like behaviors. The initial failure to find any significant effects could be attributed to a small sample size and an altered methodology. The authors also suggested that the efficacy of FAAH knockout or inhibition may be dependent on the stress levels associated with the environmental conditions. A number of studies reviewed here suggest that FAAH expression increases in response to early life stress or chronic stress. Other data also suggest that FAAH activity is increased in response to stressors and stress hormones [34][40][34,40]. It is possible, then, that the efficacy of FAAH knockout or inhibition is dependent on the severity of stress the animal experiences and the extent to which FAAH gene expression or protein level is increased. There is a large consensus across animal studies that FAAH inhibition is an effective method of reducing depressive-like behavior. This is a promising result, because it highlights the potential role of FAAH-inhibiting agents as alternative antidepressants. The most commonly used FAAH inhibitor was URB597, although other FAAH inhibitors (both reversible and irreversible) or dual inhibitors were also effective in reducing depressive-like behaviors. Only one study found no significant effects of FAAH inhibition by URB597; although, the authors employed microinjections of URB597 (0.5 and 1 μg) into the dentate gyrus of the hippocampus, whereas the other studies used larger doses by way of intraperitoneal injection [55][66]. Therefore, it is possible that the doses used in this study were not sufficient to inhibit FAAH activity and that a more widespread inhibition of FAAH activity in other brain regions in the neural circuitry of depression, such as the PFC, is required to rescue depressive phenotypes.
Notably, there appears to be dose-dependent effects with respect to the use of FAAH inhibitors. For example, Adamczyk and colleagues found that 0.1 and 0.3 mg/kg of URB597, but not 0.03 mg/kg, was sufficient in eliciting a behavioral response [56]. Similarly, Gobbi and colleagues found that 0.1 mg/kg, but not lower doses, was necessary for reducing depressive-like behaviors for both the FST and TST [54][60]. It is important to establish effective doses of each class of FAAH inhibitors for both future preclinical and clinical studies. For example, the effective doses for URB597 and URB694 evidently are 0.1–0.3 mg/kg in rodents. Determining how effective doses in rodents translate to salubrious effects in such a way that depressive symptoms are significantly attenuated will be an important avenue to pursue in human clinical research. Another valuable observation is that animals treated with FAAH inhibitors did not show Δ9-THC-like effects or deficits in locomotion, which are common side effects of direct-acting CB1 agonists. Furthermore, FAAH inhibitors do not elicit reinforcing or rewarding effects, indicating that FAAH inhibitors lack addictive properties [54][60].
Third, a large number of studies found significant differences in FAAH mainly in the PFC, hippocampus and, to a lesser extent, the striatum. These regions are not only highly implicated in depression [9], but they are also regions where greater density of ECS components are typically found [7]. Identifying the brain regions where ECS dysregulation is occurring is important in order to make connections with depressive symptoms. The PFC has a vital role in a myriad of cognitive functions including decision making, attention, motivation, problem solving as well as emotion and affect regulation [8][9][57][8,9,79]. Aberration in normal ECS signaling in the PFC could be related to functional impairments in MDD, such as inability to pay attention, lack of motivation, sad mood, or difficultly making daily decisions [8]. For example, Fagundo et al. showed that an increase in AEA levels is associated with improved decision making and cognitive flexibility in healthy female participants [58][80]. Therefore, the PFC is an important brain region of interest with respect to ECS dysregulation. In fact, the PFC is a prominent target for other treatment modalities, such as brain stimulation techniques, in MDD patients [59][81]. The hippocampus is also an important region in MDD as it is vital for learning, memory, and neuroplasticity [9]. The hippocampus is known to be affected in depression and is connected to emotion-related brain regions such as the PFC and amygdala [9]. Interestingly, greater connectivity between fronto-limbic regions has been associated with greater depressive severity [60][82]. It is widely accepted that patients with MDD have smaller hippocampal volumes and decreased hippocampal activity [61][62][83,84]. Moreover, stress can result in elevated glucocorticoids, which act on glucocorticoid receptors in the hippocampus leading to hippocampal atrophy [63][85]. Functionally, this could result in issues with autobiographical memory and negative emotion [64][53]. Other data suggest that CB1 receptor activation and endogenous release of AEA modulate memory consolidation [65][66][86,87]. Lastly, dysregulation of the ECS in the striatum could correlate with anhedonia in depression; in fact, a number of the studies reviewed here demonstrated this phenomenon using the sucrose preference test [67][88]. Disruption to dopaminergic signaling and decreased reward network connections have been associated with greater depression severity [68][89]. Functionally, these findings may translate to lack of response to rewarding or positive stimuli in MDD patients. Dysfunction in the ECS and aberrations in normal CB1 receptor signaling in these key brain regions, combined with crosstalk with other neurotransmitter systems, could play a significant role in depressive symptomology. Identifying the brain regions where ECS dysfunction has the most profound impact can help identify brain–behavior relationships with respect to specific symptoms of MDD. Therefore, the PFC, hippocampus and striatum should be considered treatment targets in clinical studies and key brain regions of interest in future neuroimaging studies in MDD patients.
Lastly, a number of studies assessed sex differences with respect to FAAH. While two studies found significant sex differences in FAAH, three studies found no differences in FAAH between the sexes [69][70][71][72][73][74][49,50,62,65,67,68]. Evidently, the data on sex differences in FAAH are mixed, which could be due to differences in the animal model or species strain. Additionally, the majority of studies on FAAH in animal models of depression used male mice and rats; this is the case in most animal studies largely due to the assumption that female estrous cycles may confound the effects of experimental manipulations [75][90]. However, there is a nearly 2-fold greater prevalence of MDD in women compared to men [76][91]. Therefore, future studies should employ female animals in preclinical studies to allow for the generalization of results involving male animal studies and to further investigate sex differences in the ECS, particularly FAAH expression. Uncovering such differences will be important to consider when targeting FAAH in clinical trials, since it is known that there are sex differences in response to current antidepressant treatments [77][92].
FAAH is a promising therapeutic target for many disorders of the CNS. Numerous potent and selective FAAH inhibitors have been developed for clinical trials in both healthy and patient populations. To our knowledge, there have only been two clinical trials evaluating FAAH inhibitors in MDD to date. One is a Phase II trial (NCT00822744, 2008-001718-26) that evaluated the efficacy of SSR411298 in elderly patients with MDD. SSR411298 did not show any significant improvement in depressive severity as measured by the Hamilton Depression Rating Scale [78][93]. Although at first glance this result seems unfavorable, FAAH inhibition could be potentially more effective in a younger population, since MDD differs considerably between younger and older adults [79][94]. The second clinical trial (NCT02498392, 2015_002007_29) evaluated the efficacy of JNJ-42165279 in participants with MDD with Anxious Distress. No results have been published to date. Therefore, more clinical trials of safe and effective FAAH inhibitors are needed in MDD.
3. Conclusions
In conclusion, the current animal models of depression show that FAAH gene expression and protein levels are dysregulated in the brain; specifically, there is increased FAAH gene expression and protein activity in the PFC, hippocampus and striatum, which correlates with the depressive phenotype. Moreover, FAAH-inhibiting agents attenuate depressive symptoms, without the side effects of direct-acting CB1 agonists. Even though results of the limited clinical trials of FAAH inhibitors in MDD have been unfavorable or unknown, the preclinical data suggest that FAAH is a promising therapeutic target for pharmacological intervention in humans with MDD and provides opportunities for combining FAAH inhibitors with other treatment modalities, such as monoamine-elevating antidepressants, transcranial magnetic stimulation, or electroconvulsive shock therapy. Therefore, more research should be performed using neuroimaging techniques in order to identify neural circuits or neurochemistry related to FAAH in vivo, and alternative FAAH inhibitors in clinical trials.