Flavonoids, widely occurring in various plants, exert a broad spectrum of beneficial effects on
human health, and are potentially powerful therapeutic tools against cancer. Recent evidences
identified numerous natural flavonoids and their derivatives as inhibitors of HIF-1, associated with
the regulation of critical glycolytic components in cancer cells, including pyruvate kinase
M2(PKM2), lactate dehydrogenase (LDHA), glucose transporters (GLUTs), hexokinase II (HKII),
phosphofructokinase-1 (PFK-1), and pyruvate dehydrogenase kinase (PDK). Here, weauthors discuss the
results of most recent studies evaluating the impact of flavonoids on HIF-1 accompanied by the
regulation of critical enzymes contributing to the Warburg phenotype. Besides, flavonoid effects on
glucose metabolism via regulation of HIF-1 activity represent a promising avenue in cancer-related
research. At the same time, only more-in depth investigations can further elucidate the
mechanistic and clinical connections between HIF-1 and cancer metabolism.
- cancer
- Warburg effect
- HIF-1
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Despite amazing progress in understanding, diagnosis, therapy, and prevention, cancer remains one of the leading causes of death worldwide, with more than 18 million new cases detected in 2018 [1,2][1][2]. Cancer is characterized as a multistage process in which tumor cells acquire specific abilities, such as uncontrolled proliferation, avoidance of apoptosis, invasiveness, and the promotion of neovascularization [3]. The uncontrolled and rapid proliferation of tumor cells and insufficient formation of new blood vessels lead to inadequate oxygen supply to tumor tissues. Therefore, it is not unusual for developing malignant tissue to possess hypoxic and necrotic areas [4]. A cell’s adaptation to a lower level of oxygen in hypoxic regions is accompanied by the activation of several survival pathways [5]. Hypoxia-inducible factor 1 (HIF-1) is a crucial transcription factor responsible for the regulation of hypoxic responses. Recent evidence has revealed that an elevated level of HIF-1 could act as a prognostic marker associated with metastasis, angiogenesis, development of chemo/radioresistance, and overall poor prognosis of cancer patients [6,7][6][7]. Metabolic reprogramming, leading to the switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis, is crucial for cell adaptation to a hypoxic environment. The favoring of aerobic glycolysis over OXPHOS (known as the Warburg effect), even at normal oxygen levels, is frequently observed in many solid tumors [8].
The stabilization of HIF-1 independently of hypoxia could explain glycolysis’s acceleration under normoxic conditions. Experimental studies have recently suggested a critical role of HIF-1 in regulating critical glycolytic proteins that contribute to the Warburg phenotype; thus, HIF-1 represents a potential target against metabolic reprogramming in cancer cells [9]. Due to its significant cancer development role, several HIF-1 inhibitors were recently clinically trailed. However, further studies on HIF-1 are necessary to provide novel therapeutic tools to inhibit its activity. Flavonoids, a class of naturally occurring plant-derived compounds, exert numerous beneficial human health attributes [10]. Flavonoids modulate various signaling pathways associated with cancer initiation, promotion, and progression, both in vitro and in vivo [4,11–13][4][11][12][13]. The effects of flavonoids on the regulatory cascade connected to HIF-1 and glucose metabolism constitute a promising way to inhibit metabolic reprogramming via the regulation of HIF-1 activity, as well as critical components of glycolysis. Therefore, in this review, we provide a comprehensive discussion of recent studies evaluating the inhibitory effects of flavonoids on HIF-1 and proteins directly contributing to the Warburg effect. The anticancer effectiveness of dietary phenols supports their application in preclinical as well as clinical research, but several complications associated with bioavailability and safety must be overcome to eliminate flavonoids’ side effects. Although flavonoids, either independently or combined with conventional therapies, could act as powerful therapeutic tools targeting cancer, further mechanistic evaluation and identifying individuals who would benefit from flavonoid-based approaches can provide hope for cancer patients.
1.1. Aim of the Study
This comprehensive review discusses the anticancer effects of plant phenolic compounds, known as flavonoids, through the targeting of HIF-1 and critical enzymes contributing to the Warburg effect. Connections between HIF-1 and metabolic reprogramming play a crucial role in cancer progression. The core of presented paper summarizes the current knowledge about the in vitro and in vivo efficacy of flavonoids against aerobic glycolysis and HIF-1 activity. Despite the lack of clinical evidence, we emphasize the possibility of introducing flavonoids (targeting HIF-1) to the clinical research considering predictive, preventive, and/or personalized medicine.
1.2. Source of the Data
The presented data were obtained from biomedical literature through the use of “hypoxia“ and “HIF-1“ or “ Warburg effect“ or “ aerobic glycolysis“ or “flavonoids“ or “flavonols“ or “chalcones“ or “anthocyanidins“ or “flavanols“ or “flavones“ or “isoflavonoids“ or “flavanones“ as either keywords or medical subject heading (MeSH) terms in searches of the PubMed database. We focused on recent publications from the last five years (2016–2020).
2. Hypoxic Conditions
Hypoxia is defined as oxygen deficiency that results in inadequate tissue oxygenation. A low oxygen level is a characteristic feature of cancer tissue. Rapid tumor growth leads to a reduced oxygen supply to specific cancer tissue areas due to insufficient vasculature development [9]. Beyond hypoxia’s role in the neovascularization necessary for the adaptation of cancer cells to oxygen and nutrient deprivation, the hypoxic state is related to other tumor features such as metabolic alterations, prolonged cell lifespan, and changes in cell adhesion and production of the extracellular matrix [14,15][14][15]. Therefore, hypoxia is a hallmark of solid tumors and is strongly connected with poor clinical prognosis due to the development of chemoresistance, radioresistance, or more aggressive forms of the disease resulting in metastasis [16,17][16][17]. The mechanism of tumor adaptation to hypoxic conditions is mediated by hypoxia-inducible factors (HIFs), which are transcription factors targeting specific genes in response to low oxygen levels [18].
2.1. Structure of Hypoxia-Inducible Factor 1
HIF-1 plays an essential role in cellular adaptation to hypoxia. Structurally, HIF-1 is a heterodimeric protein composed of HIF-α and HIF-β subunits [19]. Subunit α is further divided into three isoforms: HIF-1α, HIF-2α, and HIF-3α [20]. It is important to note that HIF-β is a constitutively expressed subunit, while HIF-α is an oxygen-regulated component of HIF-1. The helix-loop-helix domain (bHLH), responsible for DNA binding, allows the dimerization of both α and β subunits. HIF-1α and HIF-1β are composed of Per-ARNT-Sim (PAS) domains that are essential for maintaining the heterodimerization of the α and β subunits [21]. The α subunit is characterized by the oxygen-dependent degradation (ODD) domain. Prolyl-hydroxylase-2 (PHD-2) hydroxylates the ODD domain, resulting in the proteasomal degradation of the α subunit under normoxic conditions [22,23][22][23]. Other significant domains of HIF-1α include the N-terminal transactivational domain (N-TAD), which is essential for the activation of HIF-1 target genes, and the C-terminal domain (C-TAD). The C-TAD domain interacts with the C-TAD binding protein (CBP) and P300 to regulate the transcription of HIF-1α under hypoxic conditions [5,24][5][24]. Figure 1 summarizes the structures of both HIF-1α and HIF-1β subunits with their specific domains.
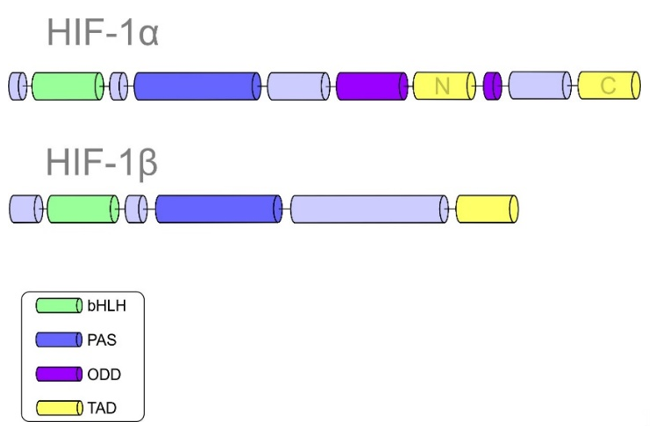
Figure 1. Domain structures of hypoxia-inducible factor 1 (HIF-1α) and HIF-1β.
2.2. Regulation of HIF-1
As mentioned above, HIF-1β is a constitutively expressed subunit, while different pathways can regulate the expression of HIF-1α. HIF-1α is degraded via the ubiquitin-proteasome pathway under normoxic conditions. Prolyl hydroxylases (PHDs) have an essential role in hydroxylating HIF-1α at its proline residues in an oxygen-dependent manner [25]. The hydroxylation of proline residues localized on the α subunit leads to ubiquitination mediated by von Hippel-Lindau suppressor (pVHL), which contains E3 ubiquitin ligase, and subsequent degradation of HIF-1α in the proteasome [26]. Importantly, PHD is inhibited, and HIF-1α is therefore stabilized under hypoxic conditions. The second way of oxygen-dependent regulation of HIF-1α is a cooperation with factor inhibiting HIF1 (FIH). FIH affects the stability of HIF-1α via hydroxylation of its asparagine residues. This interaction causes the inhibition of transcriptional coactivator recruitment [27]. Moreover, FIH and PHD are dependent on the intracellular oxygen concentration, and both require cooperation with co-factors, including 2OG (α-ketoglutarate) and Fe2+ ions. In the absence of the mentioned co-factors, HIF-1α is active even under normoxic conditions [28]. Interestingly, calcium-mediated regulation of HIF-1α is another way to modulate the activity of transcription factors. Recent evidence shows a strong correlation between disequilibrium in intracellular calcium homeostasis and HIF-1 activity [29]. For instance, calcium affects the dimerization of the receptor of activated protein C kinase (RACK1), resulting in the regulation of HIF-1α [30]. Additionally, the oxygen-independent manner of HIF-1 regulation is associated with various signaling pathways [31]. HIF-1α is affected by the ERK/MAPK [32], PI3K/AKT/mTOR [33], and JAK/STAT [29] signaling cascades. E3 ubiquitin ligase Mdm2 has an essential role in the regulation of transcription factors, including HIF-1α and p53. The interplay between p53 and HIF-1α was documented in normoxia when p53 binds to HIF-1α, leading to proteasomal degradation via Mdm2. Loss of or mutations in the tumor suppressor p53 inhibit Mdm2-mediated proteasomal degradation of HIF-1α [34]. Besides, heat shock chaperone protein 90 (HSP-90) is responsible for the stabilization of HIF-1α via conformational changes in its structure [35]. Furthermore, many studies described an important role of epigenetic mechanisms in the regulation of HIF-1 stability and activity [36–38][36][37][38]. Enzymes associated with epigenetic machinery in cells include methyltransferases, acetyltransferases, ubiquitin E3 ligases, and protein kinases that act as epigenetic writers. These enzymes can add epigenetic marks onto RNA, DNA, or histones [36,39][36][39]. Histone acetyltransferases, including c300/CBP and p300/CBP-associated factor (PCAF), promote HIF-1α stability. On the other hand, acetyltransferase arrest-defective-1 (ARD1) induces the acetylation of lysine residues and consequent ubiquitination of the HIF-1α subunit [40]. In the case of methyltransferases, methyltransferase SET7/9 is a regulator of HIF-1α stability [38]. Moreover, the HIF-α regulatory capacity of methyltransferases (PRMT1, PRMT5, and PRMT9) was detected at different levels [36,41,42][36][41][42]. Epigenetic erasers (demethylases and deacetylases) are enzymes responsible for removing specific epigenetic marks (such as methyl groups from chromatin) and, similarly to epigenetic writers, participate in chromatin remodeling and gene regulation. For instance, histone deacetylases (HDACs) (HDAC1, -2, -3, -4, -6) increase stability of HIF-1α protein [43–46][43][44][45][46]. Furthermore, HDAC7 interacts with HIF-1α and p300/CBP, and thereby promotes HIF-1 transcriptional activity. Furthermore, lysine-specific demethylase 1 (LSD1) promotes the increased stability of HIF-1α by suppressing RACK1-mediated HIF-1α degradation [38]. DNA methylation also modulates HIF-1α stability and activity. Hypermethylation of the VHL promoter region leads to constitutive activation of HIF-1α [47,48][47][48]. Numerous studies have observed a connection between HIF and non-coding RNAs. Hypoxia can enhance the expression of non-coding RNAs that can modulate HIF expression and stability. MicroRNAs, a group of small non-coding RNAs, regulate the expression of target genes at the post-transcriptional level [49]. Recent evidence suggests interplay between HIF-1α and miR-33b, miR-338-3p, miR-138, miR-576-3p, miR-143-3p, and miR-20b [50–55][50][51][52][53][54][55]. Interestingly, the regulatory role of long non-coding RNA (lncRNA) was detected in gallbladder cancer. The lncRNA LINC00152 was identified as an oncogene due to its role as a miR sponge for miR-138 targeting HIF-1α to promote carcinogenesis [56]. Similarly, the oncogenic character of lncRNAs acting as competing endogenous RNAs (ceRNAs) associated with HIF-1α was documented by HOX transcript antisense RNA (HOTAIR) (suppression of miR-217), nuclear paraspeckle assembly transcript 1 (NEAT1) (suppression of miR-186-5p), and PVT1 (suppression of miR-186) [57–59][57][58][59].
2.3. A Brief Introduction to the Warburg Phenotype
Alterations in tumor cell metabolism are a fundamental aspect of cancer compared to non-malignant cells [20]. In healthy cells, glucose is metabolized to pyruvate through the glycolytic cascade and subsequently oxidized to CO2 in mitochondria via oxidative phosphorylation. This metabolic cascade generates 36 molecules of ATP per molecule of glucose. The described pathway occurs under the normoxic condition of healthy cells [60]. During hypoxia, mitochondrial OXPHOS is restricted, leading to pyruvate conversion into lactate in a process called aerobic glycolysis [61]; notably, the overall yield of aerobic glycolysis is only 2 ATP per molecule of glucose.
Interestingly, the preference for aerobic glycolysis even under normoxic conditions and with fully functioning mitochondria is observed across different cancer types [62]. This phenomenon of metabolic reprogramming in cancer cells was first described almost 100 years ago by Otto Warburg [62]. The observation revealed that cancer cells receive enormous amounts of glucose compared to surrounding non-malignant tissue. The repression of cancer cell respiration and the enhancement of glucose fermentation into lactate were also identified [11,63][11][63]. Recent evidence has revealed the specific molecular mechanisms responsible for switching from OXPHOS to aerobic glycolysis. Alterations in molecular pathways, including PI3K/Akt/mTOR, specific genes such as cMYC and p53, and changes in epigenetic machinery directly participate in the regulation of cancer metabolism [11,64–67][11][64][65][66][67]. HIF-1 plays an essential role in the regulation of changes leading to the Warburg phenotype; it acts as a central regulator of glucose metabolism and cell proliferation.
2.4. Implementation of HIF-1 in the Modulation of Cancer Metabolism
As principally described in the previous section, the metabolic reprogramming of cancer cells allows the switch from OXPHOS to less efficient aerobic glycolysis, regulated by various molecular events such as the HIF-1 regulatory pathway (Figure 2) [11,68][11][68]. Dimerization of both HIF subunits (α and β) initiates the transcription of genes associated with increased glucose uptake and lactate production. Heterodimer HIF-1 binds to the hypoxia-response element (HRE) and begins expressing hypoxia-responsive genes [8,69][8][69]. Glucose transporters (GLUTs) are responsible for transporting glucose into the cells, and hypoxia leads to the upregulation of GLUT1 and GLUT3 [70]. Overexpression of GLUTs guarantees enough glucose for the cancer cells. Correlations between elevated HIF-1 activity and the overexpression of GLUT1 and GLUT3 were observed in glioma [71] and pancreatic neuroendocrine tumors, respectively [72]. Pyruvate kinase (PK), an enzyme responsible for converting phosphoenolpyruvate (PEP) into pyruvate, comprises four isoforms (PKL, PKR, PKM1, and PKM2). Notably, the PKM2 isoform is preferred in cancer cells, while PKM2 gene transcription is modulated by HIF-1 [73]. PKM2 acts as a coactivator via interaction with the HIF-1α subunit due to oxygen deprivation or oncogene activation [74]. This interaction promotes the transactivation of target genes regulated by HIF-1 [75]. Hexokinase (HK), another glycolytic enzyme associated with enhanced glucose metabolism, is often upregulated in poorly differentiated and highly proliferated neoplastic tissues [76]. Recently, the connections between aberrantly triggered nuclear factor-κB (NF-κB), HIF-1, and hexokinase II (HKII) expression were observed in B-cell lymphoma, while NF-kB inhibitors suppressed HIF-1 and HKII [77]. Similarly, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an enzyme that catalyzes the conversion of glyceraldehyde-3-phosphate to 1,3-biphosphoglycerate. Notably, this enzyme is often upregulated in cancer, as demonstrated in colorectal [78] and colon tumors, [79] and melanoma [80]. Deeper analysis of GADPH also revealed an association between the enzyme’s activity and HIF-1 expression in breast cancer [81]. Interestingly, the transcription of genes associated with the Warburg phenotype, including GLUT1, phosphofructokinase (PFK), enolase-1 (ENO1), PKM, aldolase A (ALDOA), and phosphoglycerate kinase-1 (PGK1), is regulated by the SIX1 transcription factor through cooperation with the histone acetyltransferases HBO1 and AIB1. Current evidence describes an essential role of miR-548a via the suppression of SIX1. Additionally, elevated HIF-1α levels are associated with decreased miR-548a, and thus enhance glycolysis and tumor growth [82].
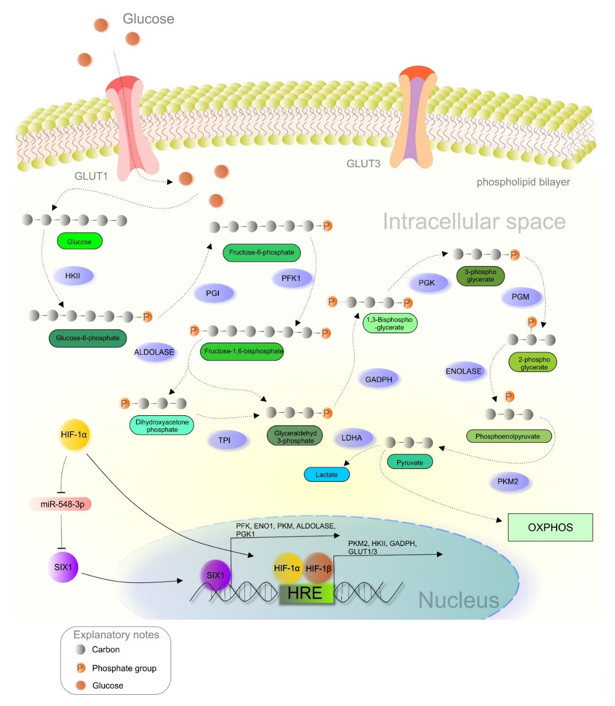
Figure 2. HIF-1α-mediated crosstalk between hypoxia and glucose metabolism in a cancer cell. Abbreviations: HKII, hexokinase II; PGI, phosphoglucose isomerase; PFK1, phosphofructokinase; TPI, triosephosphate isomerase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; PKM2, pyruvate kinase M2; LDH, lactate dehydrogenase; ENO1, enolase 1; OXPHOS, oxidative phosphorylation; HRE, hypoxia-response elements; HIF-1α/1β, hypoxia-inducible factor 1α/1β; GLUT1/3, glucose transporter 1/3.
2.5. Therapeutic Interventions Based on HIF-1 Regulation: Current Status and Future Directions
As noted above, the HIF-1 pathway modulates the expression of numerous genes involved in cancer progression and the development of resistance to different treatments [83]. Therefore, HIF, particularly its subunits HIF-1α and HIF-2α, represents a potential target of novel and potentially clinically viable oncologic interventions [84]. Several HIF inhibitors were documented to target cancer in preclinical and clinical trials. Based on their mechanisms of HIF repression, therapeutic agents are classified as factors regulating gene expression, factors modulating protein synthesis, agents modulating cascades associated with protein dimerization and accumulation, or inhibitors responsible for DNA binding and the transcriptional activity of HIF-1.
EZN-2968, an antisense oligonucleotide targeting HIF-1α mRNA, modulates HIF-1 activity at a transcriptional level; it was clinically trailed (NCT01120288, NCT02564614) [85]. Similarly, topotecan (a topoisomerase 1 inhibitor) demonstrated activity against the hypoxic phenotype by inhibiting HIF-1α mRNA expression [86,87][86][87]. Inhibitors of HIF-1α and HIF-2α protein synthesis include 2-methoxyestradiol, which affects protein synthesis in lung cancer cells [88], and KC7F2, which markedly suppresses the synthesis of HIF-1α in glioma, prostate, and breast cancers [89,90][89][90]. Another possible way to modulate HIF-1 activity is the disruption of protein stabilization and accumulation. Hsp90 inhibitors, such as geldanamycin, tanespimycin, and alvespimycin, degrade HIF-1α through a VHL-independent proteasomal mechanism [91–93][91][92][93]. Moreover, histone deacetylase inhibitors (HDACi), such as vorinostat exerted anti-hypoxic activity by degrading the HIF-1α subunit in liver cancer-derived cells [94]. Disruption of HIF-1α dimerization represents a promising way to inhibit its transcriptional activity. Recent evidence revealed an essential role of a cyclic peptide (cyclo-CLLFVY) via inhibition of HIF-1α heterodimerization [19,95][19][95].
Acriflavine showed potential as a therapeutic tool that disrupts HIF-1α dimerization [96]. Interestingly, doxorubicin and daunorubicin, well known chemotherapeutic agents, also exerted hypoxia-modulating abilities by inhibiting the binding of HIF-1 to its target (HRE) sequences [97]. Finally, epidithiodiketopiperazine chetomin inhibited the transcriptional activity of HIF-1 by targeting the HIF-1α/p300 complex in multiple myeloma cell lines [98].
Table 1 provides an overview of inhibitors that modulate HIF-1 activity at different levels of action. As was briefly analyzed in this section, inhibitors of HIF-1 are widely tested in preclinical and clinical research. Their multilevel mechanisms of action show novel opportunities in therapy and open a hidden chamber of hypoxia’s molecular secrets for a more profound understanding of the hypoxia-associated cascade in cancer. Further research in this sphere can bring well-deserved rewards in the form of effective therapeutic interventions without side effects for cancer patients.
Table 1. An overview of HIF-1 inhibitors.
|
Agents. |
Effect on HIF-1 |
References |
|
EZN-2968 |
Regulation of HIF-1α mRNA expression |
[85] |
|
Topotecan |
Regulation of HIF-1α mRNA expression |
|
|
2-Methoxyestradiol |
Modulation of HIF-1α and HIF-2α protein synthesis |
|
|
KC7F2 |
Modulation of HIF-1α protein synthesis |
|
|
Geldanamycin, tanespimycin, and alvespimycin |
Disruption of HIF-1α protein stabilization and accumulation |
|
|
Vorinostat (HDACi) |
Disruption of HIF-1α protein stabilization and accumulation |
[94] |
|
Cyclo-CLLFVY |
Inhibition of HIF-1 α heterodimerization |
|
|
Acriflavine |
Inhibition of HIF-1α heterodimerization |
[96] |
|
Doxorubicin, daunorubicin |
Inhibition of binding of HIF-1 to its target gene sequences |
[97] |
|
Chetomin |
Inhibition of the HIF-1 transcriptional activity by targeting HIF-1α/p300 |
[98] |
|
Agents. |
Effect on HIF-1 |
References |
|
EZN-2968 |
Regulation of HIF-1α mRNA expression |
[85] |
|
Topotecan |
Regulation of HIF-1α mRNA expression |
[86,87] |
|
2-Methoxyestradiol |
Modulation of HIF-1α and HIF-2α protein synthesis |
[88] |
|
KC7F2 |
Modulation of HIF-1α protein synthesis |
[89,90] |
|
Geldanamycin, tanespimycin, and alvespimycin |
Disruption of HIF-1α protein stabilization and accumulation |
[91–93] |
|
Vorinostat (HDACi) |
Disruption of HIF-1α protein stabilization and accumulation |
[94] |
|
Cyclo-CLLFVY |
Inhibition of HIF-1 α heterodimerization |
[19,95] |
|
Acriflavine |
Inhibition of HIF-1α heterodimerization |
[96] |
|
Doxorubicin, daunorubicin |
Inhibition of binding of HIF-1 to its target gene sequences |
[97] |
|
Chetomin |
Inhibition of the HIF-1 transcriptional activity by targeting HIF-1α/p300 |
[98] |
Abbreviations: HIF-1, hypoxia-inducible factor 1; HIF-1α/2α, hypoxia-inducible factor 1α/2α; HDCAi, histone deacetylase inhibitor
References
- Biemar, F.; Foti, M. Global Progress against Cancer—Challenges and Opportunities. Cancer Biol. Med. 2013, 10, 183–186, doi:10.7497/j.issn.2095-3941.2013.04.001.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424, doi:10.3322/caac.21492.
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of Survival, Proliferation, Invasion, Angiogenesis, and Metastasis of Tumor Cells Through Modulation of Inflammatory Pathways by Nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434, doi:10.1007/s10555-010-9235-2.
- Deng, X.; Peng, Y.; Zhao, J.; Lei, X.; Zheng, X.; Xie, Z.; Tang, G. Anticancer Activity of Natural Flavonoids: Inhibition of HIF-1α Signaling Pathway. Org. Chem. 2020, 23, 2945–2959, doi:10.2174/1385272823666191203122030.
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389, doi:10.1016/j.apsb.2015.05.007.
- Jögi, A.; Ehinger, A.; Hartman, L.; Alkner, S. Expression of HIF-1α Is Related to a Poor Prognosis and Tamoxifen Resistance in Contralateral Breast Cancer. PLoS ONE 2019, 14, e0226150, doi:10.1371/journal.pone.0226150.
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review Available online: https://www.eurekaselect.com/167204/article (accessed on 4 November 2020).
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20020238.
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Cell Dev. Biol. 2019, 7, doi:10.3389/fcell.2019.00004.
- Kozłowska, A.; Szostak-Wegierek, D. Flavonoids—Food Sources and Health Benefits. Państwowego Zakładu Hig. 2014, 65, 79–85.
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg Phenotype—Concepts of Predictive, Preventive and Personalised Medicine to Cut the Gordian Knot of Cancer Cell Metabolism. EPMA J. 2020, 11, 377–398, doi:10.1007/s13167-020-00217-y.
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, doi:10.3390/cancers12061498.
- Liskova, A.; Koklesova, L.; Samec, M.; Varghese, E.; Abotaleb, M.; Samuel, S.M.; Smejkal, K.; Biringer, K.; Petras, M.; Blahutova, D.; et al. Implications of Flavonoids as Potential Modulators of Cancer Neovascularity. Cancer Res. Clin. Oncol. 2020, doi:10.1007/s00432-020-03383-8.
- Carnero, A.; Lleonart, M. The Hypoxic Microenvironment: A Determinant of Cancer Stem Cell Evolution. Cell 2016, 1, 96–105, doi:10.1002/icl3.1039.
- Finger, E.C.; Giaccia, A.J. Hypoxia, Inflammation, and the Tumor Microenvironment in Metastatic Disease. Cancer Metastasis Rev. 2010, 29, 285–293, doi:10.1007/s10555-010-9224-5.
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of Hypoxic Tumor Microenvironment and Tumor Cell Plasticity on the Expression of Immune Checkpoints. Cancer Lett. 2019, 458, 13–20, doi:10.1016/j.canlet.2019.05.021.
- Rankin, E.B.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304, doi:10.1016/j.trecan.2016.05.006.
- Balamurugan, K. HIF-1 at the Crossroads of Hypoxia, Inflammation, and Cancer. J. Cancer 2016, 138, 1058–1066, doi:10.1002/ijc.29519.
- Burslem, G.M.; Kyle, H.F.; Nelson, A.; Edwards, T.A.; Wilson, A.J. Hypoxia Inducible Factor (HIF) as a Model for Studying Inhibition of Protein-Protein Interactions. Sci. 2017, 8, 4188–4202, doi:10.1039/C7SC00388A.
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Biol. Rep. 2015, 42, 841–851, doi:10.1007/s11033-015-3858-x.
- Feng, Z.; Zou, X.; Chen, Y.; Wang, H.; Duan, Y.; Bruick, R.K. Modulation of HIF-2α PAS-B Domain Contributes to Physiological Responses. Natl. Acad. Sci. USA 2018, 115, 13240–13245, doi:10.1073/pnas.1810897115.
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60.
- Zhao, L.; Liu, Z.; Yang, F.; Zhang, Y.; Xue, Y.; Miao, H.; Liao, X.; Huang, H.; Li, G. Intrabody against Prolyl Hydroxylase 2 Promotes Angiogenesis by Stabilizing Hypoxia-Inducible Factor-1α. Rep. 2019, 9, 11861, doi:10.1038/s41598-019-47891-1.
- Guo, Y.; Xiao, Z.; Yang, L.; Gao, Y.; Zhu, Q.; Hu, L.; Huang, D.; Xu, Q. Hypoxia‑Inducible Factors in Hepatocellular Carcinoma (Review). Rep. 2020, 43, 3–15, doi:10.3892/or.2019.7397.
- Wu, L.-Y.; He, Y.-L.; Zhu, L.-L. Possible Role of PHD Inhibitors as Hypoxia-Mimicking Agents in the Maintenance of Neural Stem Cells’ Self-Renewal Properties. Cell Dev. Biol. 2018, 6, doi:10.3389/fcell.2018.00169.
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-Hydroxylated Hypoxia-Inducible Factor 1α (HIF-1α) Upregulation in Human Tumours. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0088955.
- Wang, E.; Zhang, C.; Polavaram, N.; Liu, F.; Wu, G.; Schroeder, M.A.; Lau, J.S.; Mukhopadhyay, D.; Jiang, S.-W.; O’Neill, B.P.; et al. The Role of Factor Inhibiting HIF (FIH-1) in Inhibiting HIF-1 Transcriptional Activity in Glioblastoma Multiforme. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0086102.
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20133278.
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Oncol. 2017, 7, doi:10.3389/fonc.2017.00286.
- Liu, Y.V.; Hubbi, M.E.; Pan, F.; McDonald, K.R.; Mansharamani, M.; Cole, R.N.; Liu, J.O.; Semenza, G.L. Calcineurin Promotes Hypoxia-Inducible Factor 1α Expression by Dephosphorylating RACK1 and Blocking RACK1 Dimerization. Biol. Chem. 2007, 282, 37064–37073, doi:10.1074/jbc.M705015200.
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of Hypoxia-Inducible Factor and Metabolic Pathways: Possible Targets of Cancer. Cell Biosci. 2017, 7, doi:10.1186/s13578-017-0190-2.
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and P38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202, doi:10.1158/0008-5472.CAN-18-0270.
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 Signaling Pathway in Hypoxia-Ischemia. Med. Rep. 2018, 18, 3547–3554, doi:10.3892/mmr.2018.9375.
- Robertson, E.D.; Semenchenko, K.; Wasylyk, B. Crosstalk between Mdm2, P53 and HIF1-α: Distinct Responses to Oxygen Stress and Implications for Tumour Hypoxia. Biochem. 2014, 85, 199–214, doi:10.1007/978-94-017-9211-0_11.
- Kataria, N.; Kerr, B.; Zaiter, S.S.; McAlpine, S.; Cook, K.M. C-Terminal HSP90 Inhibitors Block the HSP90:HIF-1α Interaction and Inhibit the Cellular Hypoxic Response. bioRxiv 2019, 521989, doi:10.1101/521989.
- Luo, W.; Wang, Y. Epigenetic Regulators: Multifunctional Proteins Modulating Hypoxia-Inducible Factor-α Protein Stability and Activity. Mol. Life Sci. 2018, 75, 1043–1056, doi:10.1007/s00018-017-2684-9.
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia Induces Rapid Changes to Histone Methylation and Reprograms Chromatin. Science 2019, 363, 1222–1226, doi:10.1126/science.aau5870.
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-Dependent Regulation of HIF-1α Stability Restricts Retinal and Tumour Angiogenesis. Commun. 2016, 7, 10347, doi:10.1038/ncomms10347.
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298, doi:10.1016/j.cmet.2017.10.005.
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Rev. Biochem. Mol. Biol. 2014, 49, 1–15, doi:10.3109/10409238.2013.838205.
- Lafleur, V.N.; Richard, S.; Richard, D.E. Transcriptional Repression of Hypoxia-Inducible Factor-1 (HIF-1) by the Protein Arginine Methyltransferase PRMT1. Biol. Cell 2014, 25, 925–935, doi:10.1091/mbc.e13-07-0423.
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 Enhances Tumorigenicity and Glycolysis in Pancreatic Cancer via the FBW7/CMyc Axis. Cell Commun. Signal. 2019, 17, 30, doi:10.1186/s12964-019-0344-4.
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs Mediate HIF-1α Stability Through PHD2-Dependent Mechanism While HDAC6, a Class IIb Member, Promotes HIF-1α Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. Bone Miner. Res. 2016, 31, 1287–1299, doi:10.1002/jbmr.2787.
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 Modulate Cell Invasion and Migration in Clear Cell Renal Cell Carcinoma. BMC Cancer 2016, 16, 617, doi:10.1186/s12885-016-2604-7.
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 Protein Regulates HIF1α Protein Lysine Acetylation and Cancer Cell Response to Hypoxia. Biol. Chem. 2011, 286, 38095–38102, doi:10.1074/jbc.M111.257055.
- Chen, S.; Sang, N. Histone Deacetylase Inhibitors: The Epigenetic Therapeutics That Repress Hypoxia-Inducible Factors. Available online: https://www.hindawi.com/journals/bmri/2011/197946/ (accessed on 11 October 2020).
- Robinson, C.M.; Lefebvre, F.; Poon, B.P.; Bousard, A.; Fan, X.; Lathrop, M.; Tost, J.; Kim, W.Y.; Riazalhosseini, Y.; Ohh, M. Consequences of VHL Loss on Global DNA Methylome. Rep. 2018, 8, doi:10.1038/s41598-018-21524-5.
- Schmitt, A.M.; Schmid, S.; Rudolph, T.; Anlauf, M.; Prinz, C.; Klöppel, G.; Moch, H.; Heitz, P.U.; Komminoth, P.; Perren, A. VHL Inactivation Is an Important Pathway for the Development of Malignant Sporadic Pancreatic Endocrine Tumors. Relat. Cancer 2009, 16, 1219–1227, doi:10.1677/ERC-08-0297.
- Holubekova, V.; Mendelova, A.; Jasek, K.; Mersakova, S.; Zubor, P.; Lasabova, Z. Epigenetic Regulation by DNA Methylation and MiRNA Molecules in Cancer. Future Oncol. 2017, 13, 2217–2222, doi:10.2217/fon-2017-0363.
- Zhou, Y.; Yang, C.; Wang, K.; Liu, X.; Liu, Q. MicroRNA-33b Inhibits the Proliferation and Migration of Osteosarcoma Cells via Targeting Hypoxia-Inducible Factor-1α. Res. 2017, 25, 397–405, doi:10.3727/096504016X14743337535446.
- Xu, H.; Zhao, L.; Fang, Q.; Sun, J.; Zhang, S.; Zhan, C.; Liu, S.; Zhang, Y. MiR-338-3p Inhibits Hepatocarcinoma Cells and Sensitizes These Cells to Sorafenib by Targeting Hypoxia-Induced Factor 1α. PLoS ONE 2014, 9, e115565, doi:10.1371/journal.pone.0115565.
- Yeh, Y.-M.; Chuang, C.-M.; Chao, K.-C.; Wang, L.-H. MicroRNA-138 Suppresses Ovarian Cancer Cell Invasion and Metastasis by Targeting SOX4 and HIF-1α. J. Cancer 2013, 133, 867–878, doi:10.1002/ijc.28086.
- Hu, Q.; Liu, F.; Yan, T.; Wu, M.; Ye, M.; Shi, G.; Lv, S.; Zhu, X. MicroRNA‑576‑3p Inhibits the Migration and Proangiogenic Abilities of Hypoxia‑treated Glioma Cells Through Hypoxia‑Inducible Factor‑1α. J. Mol. Med. 2019, 43, 2387–2397, doi:10.3892/ijmm.2019.4157.
- He, M.; Zhan, M.; Chen, W.; Xu, S.; Long, M.; Shen, H.; Shi, Y.; Liu, Q.; Mohan, M.; Wang, J. MiR-143-5p Deficiency Triggers EMT and Metastasis by Targeting HIF-1α in Gallbladder Cancer. Physiol. Biochem. 2017, 42, 2078–2092, doi:10.1159/000479903.
- Xue, T.-M.; Tao, L.; Zhang, M.; Zhang, J.; Liu, X.; Chen, G.-F.; Zhu, Y.-J.; Zhang, P.-J. Clinicopathological Significance of MicroRNA-20b Expression in Hepatocellular Carcinoma and Regulation of HIF-1α and VEGF Effect on Cell Biological Behaviour. Markers 2015, 2015, 325176, doi:10.1155/2015/325176.
- Cai, Q.; Wang, Z.; Wang, S.; Weng, M.; Zhou, D.; Li, C.; Wang, J.; Chen, E.; Quan, Z. Long Non-Coding RNA LINC00152 Promotes Gallbladder Cancer Metastasis and Epithelial-Mesenchymal Transition by Regulating HIF-1α via MiR-138. Open Biol. 2017, 7, doi:10.1098/rsob.160247.
- Hong, Q.; Li, O.; Zheng, W.; Xiao, W.-Z.; Zhang, L.; Wu, D.; Cai, G.-Y.; He, J.C.; Chen, X.-M. LncRNA HOTAIR Regulates HIF-1α/AXL Signaling Through Inhibition of MiR-217 in Renal Cell Carcinoma. Cell Death Dis. 2017, 8, e2772, doi:10.1038/cddis.2017.181.
- Tan, H.; Zhao, L. LncRNA Nuclear-Enriched Abundant Transcript 1 Promotes Cell Proliferation and Invasion by Targeting MiR-186-5p/HIF-1α in Osteosarcoma. Cell. Biochem. 2019, 120, 6502–6514, doi:10.1002/jcb.27941.
- Huang, T.; Liu, H.W.; Chen, J.Q.; Wang, S.H.; Hao, L.Q.; Liu, M.; Wang, B. The Long Noncoding RNA PVT1 Functions as a Competing Endogenous RNA by Sponging MiR-186 in Gastric Cancer. Pharmacother. 2017, 88, 302–308, doi:10.1016/j.biopha.2017.01.049.
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Endocrinol. 2017, 8, doi:10.3389/fendo.2017.00279.
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. J. Radiat. Biol. 2019, 95, 912–919, doi:10.1080/09553002.2019.1589653.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218, doi:10.1016/j.tibs.2015.12.001.
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree Effects: On the Origin of Cancer Cell Energy Metabolism and of Yeast Glucose Repression. Biophys. Acta BBA Bioenerg. 2011, 1807, 568–576, doi:10.1016/j.bbabio.2010.08.010.
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Endocrinol. 2018, 9, doi:10.3389/fendo.2018.00129.
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Rev. Cancer 2020, 20, 74–88, doi:10.1038/s41568-019-0216-7.
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Genet. 2018, 9, doi:10.3389/fgene.2018.00427.
- Wilkie, M.D.; Anaam, E.A.; Lau, A.S.; Rubbi, C.P.; Jones, T.M.; Boyd, M.T.; Vlatković, N. TP53 Mutations in Head and Neck Cancer Cells Determine the Warburg Phenotypic Switch Creating Metabolic Vulnerabilities and Therapeutic Opportunities for Stratified Therapies. Cancer Lett. 2020, 478, 107–121, doi:10.1016/j.canlet.2020.02.032.
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, doi:10.3390/cancers12082252.
- Dabral, S.; Muecke, C.; Valasarajan, C.; Schmoranzer, M.; Wietelmann, A.; Semenza, G.L.; Meister, M.; Muley, T.; Seeger-Nukpezah, T.; Samakovlis, C.; et al. A RASSF1A-HIF1α Loop Drives Warburg Effect in Cancer and Pulmonary Hypertension. Commun. 2019, 10, 2130, doi:10.1038/s41467-019-10044-z.
- Sadlecki, P.; Bodnar, M.; Grabiec, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Zegarska, J.; Walentowicz-Sadlecka, M. The Role of Hypoxia-Inducible Factor-1α, Glucose Transporter-1, (GLUT-1) and Carbon Anhydrase IX in Endometrial Cancer Patients. Available online: https://www.hindawi.com/journals/bmri/2014/616850/ (accessed on 13 October 2020).
- Liu, Y.; Li, Y.; Tian, R.; Liu, W.; Fei, Z.; Long, Q.; Wang, X.; Zhang, X. The Expression and Significance of HIF-1α and GLUT-3 in Glioma. Brain Res. 2009, 1304, 149–154, doi:10.1016/j.brainres.2009.09.083.
- Fujino, M.; Aishima, S.; Shindo, K.; Oda, Y.; Morimatsu, K.; Tsutsumi, K.; Otsuka, T.; Tanaka, M.; Oda, Y. Expression of Glucose Transporter-1 Is Correlated with Hypoxia-Inducible Factor 1 (Alpha) and Malignant Potential in Pancreatic Neuroendocrine Tumors. Lett. 2016, 12, 3337–3344.
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α. JAK STAT 2012, 1, 194–196, doi:10.4161/jkst.20662.
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and Cancer: The Function of PKM2 beyond Glycolysis (Review). Lett. 2016, 11, 1980–1986, doi:10.3892/ol.2016.4168.
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnson, D.G.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction, and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 65–80, doi:10.1016/j.cmet.2014.12.005.
- Tan, V.P.; Miyamoto, S. HK2/Hexokinase-II Integrates Glycolysis and Autophagy to Confer Cellular Protection. Autophagy 2015, 11, 963–964, doi:10.1080/15548627.2015.1042195.
- Nakajima, K.; Kirito, K.; Kawashima, I.; Koshiishi, M.; Kumagai, T.; Mitsumori, T. Aberrant Activation of NF-ΚB and Hypoxia Enhances Glycolytic Enzyme Hexokinase II Expression in B-Cell Lymphoma Through Activation of HIF-1. Blood 2017, 130, 4025–4025, doi:10.1182/blood.V130.Suppl_1.4025.4025.
- Tarrado-Castellarnau, M.; Diaz-Moralli, S.; Polat, I.H.; Sanz-Pamplona, R.; Alenda, C.; Moreno, V.; Castells, A.; Cascante, M. Glyceraldehyde-3-Phosphate Dehydrogenase Is Overexpressed in Colorectal Cancer Onset. Med. Commun. 2017, 2, 6, doi:10.1186/s41231-017-0015-7.
- Liu, K.; Tang, Z.; Huang, A.; Chen, P.; Liu, P.; Yang, J.; Lu, W.; Liao, J.; Sun, Y.; Wen, S.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase Promotes Cancer Growth and Metastasis Through Upregulation of SNAIL Expression. J. Oncol. 2017, 50, 252–262, doi:10.3892/ijo.2016.3774.
- Jadhav, S.; Krynetskaia, N.; Krynetskiy, E. Cellular Response to Anticancer Drugs in Melanoma Cells: Inhibition of Glyceraldehyde 3-Phosphate Dehydrogenase Results in Cell Cycle Arrest and Increased Chemoresistance to Chemotherapeutic Agents. Cancer Res. 2007, 67, 3238–3238.
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-Regulation of Glyceraldehyde-3-Phosphate Dehydrogenase Gene Expression by HIF-1 Activity Depending on Sp1 in Hypoxic Breast Cancer Cells. Biochem. Biophys. 2011, 509, 1–8, doi:10.1016/j.abb.2011.02.011.
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7, doi:10.1016/j.ccell.2018.01.010.
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496, doi:10.3349/ymj.2017.58.3.489.
- Albadari, N.; Deng, S.; Li, W. The Transcriptional Factors HIF-1 and HIF-2 and Their Novel Inhibitors in Cancer Therapy. Expert Opin. Drug Discov. 2019, 14, 667–682, doi:10.1080/17460441.2019.1613370.
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot Trial of EZN-2968, an Antisense Oligonucleotide Inhibitor of Hypoxia-Inducible Factor-1 Alpha (HIF-1α), in Patients with Refractory Solid Tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348, doi:10.1007/s00280-013-2362-z.
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic Targeting of Hypoxia and Hypoxia-Inducible Factors in Cancer. Ther. 2016, 164, 152–169, doi:10.1016/j.pharmthera.2016.04.009.
- Beppu, K.; Nakamura, K.; Linehan, W.M.; Rapisarda, A.; Thiele, C.J. Topotecan Blocks Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor Expression Induced by Insulin-like Growth Factor-I in Neuroblastoma Cells. Cancer Res. 2005, 65, 4775–4781, doi:10.1158/0008-5472.CAN-04-3332.
- Aquino-Gálvez, A.; González-Ávila, G.; Delgado-Tello, J.; Castillejos-López, M.; Mendoza-Milla, C.; Zúñiga, J.; Checa, M.; Maldonado-Martínez, H.A.; Trinidad-López, A.; Cisneros, J.; et al. Effects of 2-Methoxyestradiol on Apoptosis and HIF-1α and HIF-2α Expression in Lung Cancer Cells under Normoxia and Hypoxia. Rep. 2016, 35, 577–583, doi:10.3892/or.2015.4399.
- Narita, T.; Yin, S.; Gelin, C.F.; Moreno, C.S.; Yepes, M.; Nicolaou, K.C.; Van Meir, E.G. Identification of a Novel Small Molecule HIF-1α Translation Inhibitor. Cancer Res. 2009, 15, 6128–6136, doi:10.1158/1078-0432.CCR-08-3180.
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-Inducible Factor-1 and Associated Upstream and Downstream Proteins in the Pathophysiology and Management of Glioblastoma. Focus 2014, 37, E8, doi:10.3171/2014.9.FOCUS14496.
- Papale, M.; Ferretti, E.; Battaglia, G.; Bellavia, D.; Mai, A.; Tafani, M. EZH2, HIF-1, and Their Inhibitors: An Overview on Pediatric Cancers. Pediatr. 2018, 6, doi:10.3389/fped.2018.00328.
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Oncol. Rep. 2019, 21, 6, doi:10.1007/s11912-019-0752-z.
- Ibrahim, N.O.; Hahn, T.; Franke, C.; Stiehl, D.P.; Wirthner, R.; Wenger, R.H.; Katschinski, D.M. Induction of the Hypoxia-Inducible Factor System by Low Levels of Heat Shock Protein 90 Inhibitors. Cancer Res. 2005, 65, 11094–11100, doi:10.1158/0008-5472.CAN-05-1877.
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The Histone Deacetylase Inhibitor, Vorinostat, Represses Hypoxia Inducible Factor 1 Alpha Expression Through Translational Inhibition. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0106224.
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. Am. Chem. Soc. 2013, 135, 10418–10425, doi:10.1021/ja402993u.
- Mangraviti, A.; Raghavan, T.; Volpin, F.; Skuli, N.; Gullotti, D.; Zhou, J.; Asnaghi, L.; Sankey, E.; Liu, A.; Wang, Y.; et al. HIF-1α- Targeting Acriflavine Provides Long Term Survival and Radiological Tumor Response in Brain Cancer Therapy. Rep. 2017, 7, 14978, doi:10.1038/s41598-017-14990-w.
- Lee, K.; Qian, D.Z.; Rey, S.; Wei, H.; Liu, J.O.; Semenza, G.L. Anthracycline Chemotherapy Inhibits HIF-1 Transcriptional Activity and Tumor-Induced Mobilization of Circulating Angiogenic Cells. Natl. Acad. Sci. USA 2009, 106, 2353–2358, doi:10.1073/pnas.0812801106.
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, Targeting HIF-1α/P300 Complex, Exhibits Antitumour Activity in Multiple Myeloma. J. Cancer 2016, 114, 519–523, doi:10.1038/bjc.2016.20.
.
References
- Biemar, F.; Foti, M. Global Progress against Cancer—Challenges and Opportunities. Cancer Biol. Med. 2013, 10, 183–186, doi:10.7497/j.issn.2095-3941.2013.04.001.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424, doi:10.3322/caac.21492.
- Gupta, S.C.; Kim, J.H.; Prasad, S.; Aggarwal, B.B. Regulation of Survival, Proliferation, Invasion, Angiogenesis, and Metastasis of Tumor Cells Through Modulation of Inflammatory Pathways by Nutraceuticals. Cancer Metastasis Rev. 2010, 29, 405–434, doi:10.1007/s10555-010-9235-2.
- Deng, X.; Peng, Y.; Zhao, J.; Lei, X.; Zheng, X.; Xie, Z.; Tang, G. Anticancer Activity of Natural Flavonoids: Inhibition of HIF-1α Signaling Pathway. Org. Chem. 2020, 23, 2945–2959, doi:10.2174/1385272823666191203122030.
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389, doi:10.1016/j.apsb.2015.05.007.
- Jögi, A.; Ehinger, A.; Hartman, L.; Alkner, S. Expression of HIF-1α Is Related to a Poor Prognosis and Tamoxifen Resistance in Contralateral Breast Cancer. PLoS ONE 2019, 14, e0226150, doi:10.1371/journal.pone.0226150.
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review Available online: https://www.eurekaselect.com/167204/article (accessed on 4 November 2020).
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20020238.
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-Modified Cancer Cell Metabolism. Cell Dev. Biol. 2019, 7, doi:10.3389/fcell.2019.00004.
- Kozłowska, A.; Szostak-Wegierek, D. Flavonoids—Food Sources and Health Benefits. Państwowego Zakładu Hig. 2014, 65, 79–85.
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg Phenotype—Concepts of Predictive, Preventive and Personalised Medicine to Cut the Gordian Knot of Cancer Cell Metabolism. EPMA J. 2020, 11, 377–398, doi:10.1007/s13167-020-00217-y.
- Liskova, A.; Koklesova, L.; Samec, M.; Smejkal, K.; Samuel, S.M.; Varghese, E.; Abotaleb, M.; Biringer, K.; Kudela, E.; Danko, J.; et al. Flavonoids in Cancer Metastasis. Cancers 2020, 12, doi:10.3390/cancers12061498.
- Liskova, A.; Koklesova, L.; Samec, M.; Varghese, E.; Abotaleb, M.; Samuel, S.M.; Smejkal, K.; Biringer, K.; Petras, M.; Blahutova, D.; et al. Implications of Flavonoids as Potential Modulators of Cancer Neovascularity. Cancer Res. Clin. Oncol. 2020, doi:10.1007/s00432-020-03383-8.
- Carnero, A.; Lleonart, M. The Hypoxic Microenvironment: A Determinant of Cancer Stem Cell Evolution. Cell 2016, 1, 96–105, doi:10.1002/icl3.1039.
- Finger, E.C.; Giaccia, A.J. Hypoxia, Inflammation, and the Tumor Microenvironment in Metastatic Disease. Cancer Metastasis Rev. 2010, 29, 285–293, doi:10.1007/s10555-010-9224-5.
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of Hypoxic Tumor Microenvironment and Tumor Cell Plasticity on the Expression of Immune Checkpoints. Cancer Lett. 2019, 458, 13–20, doi:10.1016/j.canlet.2019.05.021.
- Rankin, E.B.; Nam, J.-M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304, doi:10.1016/j.trecan.2016.05.006.
- Balamurugan, K. HIF-1 at the Crossroads of Hypoxia, Inflammation, and Cancer. J. Cancer 2016, 138, 1058–1066, doi:10.1002/ijc.29519.
- Burslem, G.M.; Kyle, H.F.; Nelson, A.; Edwards, T.A.; Wilson, A.J. Hypoxia Inducible Factor (HIF) as a Model for Studying Inhibition of Protein-Protein Interactions. Sci. 2017, 8, 4188–4202, doi:10.1039/C7SC00388A.
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Biol. Rep. 2015, 42, 841–851, doi:10.1007/s11033-015-3858-x.
- Feng, Z.; Zou, X.; Chen, Y.; Wang, H.; Duan, Y.; Bruick, R.K. Modulation of HIF-2α PAS-B Domain Contributes to Physiological Responses. Natl. Acad. Sci. USA 2018, 115, 13240–13245, doi:10.1073/pnas.1810897115.
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and Its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60.
- Zhao, L.; Liu, Z.; Yang, F.; Zhang, Y.; Xue, Y.; Miao, H.; Liao, X.; Huang, H.; Li, G. Intrabody against Prolyl Hydroxylase 2 Promotes Angiogenesis by Stabilizing Hypoxia-Inducible Factor-1α. Rep. 2019, 9, 11861, doi:10.1038/s41598-019-47891-1.
- Guo, Y.; Xiao, Z.; Yang, L.; Gao, Y.; Zhu, Q.; Hu, L.; Huang, D.; Xu, Q. Hypoxia‑Inducible Factors in Hepatocellular Carcinoma (Review). Rep. 2020, 43, 3–15, doi:10.3892/or.2019.7397.
- Wu, L.-Y.; He, Y.-L.; Zhu, L.-L. Possible Role of PHD Inhibitors as Hypoxia-Mimicking Agents in the Maintenance of Neural Stem Cells’ Self-Renewal Properties. Cell Dev. Biol. 2018, 6, doi:10.3389/fcell.2018.00169.
- Snell, C.E.; Turley, H.; McIntyre, A.; Li, D.; Masiero, M.; Schofield, C.J.; Gatter, K.C.; Harris, A.L.; Pezzella, F. Proline-Hydroxylated Hypoxia-Inducible Factor 1α (HIF-1α) Upregulation in Human Tumours. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0088955.
- Wang, E.; Zhang, C.; Polavaram, N.; Liu, F.; Wu, G.; Schroeder, M.A.; Lau, J.S.; Mukhopadhyay, D.; Jiang, S.-W.; O’Neill, B.P.; et al. The Role of Factor Inhibiting HIF (FIH-1) in Inhibiting HIF-1 Transcriptional Activity in Glioblastoma Multiforme. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0086102.
- Nakayama, K.; Kataoka, N. Regulation of Gene Expression under Hypoxic Conditions. J. Mol. Sci. 2019, 20, doi:10.3390/ijms20133278.
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Oncol. 2017, 7, doi:10.3389/fonc.2017.00286.
- Liu, Y.V.; Hubbi, M.E.; Pan, F.; McDonald, K.R.; Mansharamani, M.; Cole, R.N.; Liu, J.O.; Semenza, G.L. Calcineurin Promotes Hypoxia-Inducible Factor 1α Expression by Dephosphorylating RACK1 and Blocking RACK1 Dimerization. Biol. Chem. 2007, 282, 37064–37073, doi:10.1074/jbc.M705015200.
- Singh, D.; Arora, R.; Kaur, P.; Singh, B.; Mannan, R.; Arora, S. Overexpression of Hypoxia-Inducible Factor and Metabolic Pathways: Possible Targets of Cancer. Cell Biosci. 2017, 7, doi:10.1186/s13578-017-0190-2.
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and P38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202, doi:10.1158/0008-5472.CAN-18-0270.
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 Signaling Pathway in Hypoxia-Ischemia. Med. Rep. 2018, 18, 3547–3554, doi:10.3892/mmr.2018.9375.
- Robertson, E.D.; Semenchenko, K.; Wasylyk, B. Crosstalk between Mdm2, P53 and HIF1-α: Distinct Responses to Oxygen Stress and Implications for Tumour Hypoxia. Biochem. 2014, 85, 199–214, doi:10.1007/978-94-017-9211-0_11.
- Kataria, N.; Kerr, B.; Zaiter, S.S.; McAlpine, S.; Cook, K.M. C-Terminal HSP90 Inhibitors Block the HSP90:HIF-1α Interaction and Inhibit the Cellular Hypoxic Response. bioRxiv 2019, 521989, doi:10.1101/521989.
- Luo, W.; Wang, Y. Epigenetic Regulators: Multifunctional Proteins Modulating Hypoxia-Inducible Factor-α Protein Stability and Activity. Mol. Life Sci. 2018, 75, 1043–1056, doi:10.1007/s00018-017-2684-9.
- Batie, M.; Frost, J.; Frost, M.; Wilson, J.W.; Schofield, P.; Rocha, S. Hypoxia Induces Rapid Changes to Histone Methylation and Reprograms Chromatin. Science 2019, 363, 1222–1226, doi:10.1126/science.aau5870.
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-Dependent Regulation of HIF-1α Stability Restricts Retinal and Tumour Angiogenesis. Commun. 2016, 7, 10347, doi:10.1038/ncomms10347.
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298, doi:10.1016/j.cmet.2017.10.005.
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional Regulation by Hypoxia Inducible Factors. Rev. Biochem. Mol. Biol. 2014, 49, 1–15, doi:10.3109/10409238.2013.838205.
- Lafleur, V.N.; Richard, S.; Richard, D.E. Transcriptional Repression of Hypoxia-Inducible Factor-1 (HIF-1) by the Protein Arginine Methyltransferase PRMT1. Biol. Cell 2014, 25, 925–935, doi:10.1091/mbc.e13-07-0423.
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 Enhances Tumorigenicity and Glycolysis in Pancreatic Cancer via the FBW7/CMyc Axis. Cell Commun. Signal. 2019, 17, 30, doi:10.1186/s12964-019-0344-4.
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs Mediate HIF-1α Stability Through PHD2-Dependent Mechanism While HDAC6, a Class IIb Member, Promotes HIF-1α Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. Bone Miner. Res. 2016, 31, 1287–1299, doi:10.1002/jbmr.2787.
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 Modulate Cell Invasion and Migration in Clear Cell Renal Cell Carcinoma. BMC Cancer 2016, 16, 617, doi:10.1186/s12885-016-2604-7.
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 Protein Regulates HIF1α Protein Lysine Acetylation and Cancer Cell Response to Hypoxia. Biol. Chem. 2011, 286, 38095–38102, doi:10.1074/jbc.M111.257055.
- Chen, S.; Sang, N. Histone Deacetylase Inhibitors: The Epigenetic Therapeutics That Repress Hypoxia-Inducible Factors. Available online: https://www.hindawi.com/journals/bmri/2011/197946/ (accessed on 11 October 2020).
- Robinson, C.M.; Lefebvre, F.; Poon, B.P.; Bousard, A.; Fan, X.; Lathrop, M.; Tost, J.; Kim, W.Y.; Riazalhosseini, Y.; Ohh, M. Consequences of VHL Loss on Global DNA Methylome. Rep. 2018, 8, doi:10.1038/s41598-018-21524-5.
- Schmitt, A.M.; Schmid, S.; Rudolph, T.; Anlauf, M.; Prinz, C.; Klöppel, G.; Moch, H.; Heitz, P.U.; Komminoth, P.; Perren, A. VHL Inactivation Is an Important Pathway for the Development of Malignant Sporadic Pancreatic Endocrine Tumors. Relat. Cancer 2009, 16, 1219–1227, doi:10.1677/ERC-08-0297.
- Holubekova, V.; Mendelova, A.; Jasek, K.; Mersakova, S.; Zubor, P.; Lasabova, Z. Epigenetic Regulation by DNA Methylation and MiRNA Molecules in Cancer. Future Oncol. 2017, 13, 2217–2222, doi:10.2217/fon-2017-0363.
- Zhou, Y.; Yang, C.; Wang, K.; Liu, X.; Liu, Q. MicroRNA-33b Inhibits the Proliferation and Migration of Osteosarcoma Cells via Targeting Hypoxia-Inducible Factor-1α. Res. 2017, 25, 397–405, doi:10.3727/096504016X14743337535446.
- Xu, H.; Zhao, L.; Fang, Q.; Sun, J.; Zhang, S.; Zhan, C.; Liu, S.; Zhang, Y. MiR-338-3p Inhibits Hepatocarcinoma Cells and Sensitizes These Cells to Sorafenib by Targeting Hypoxia-Induced Factor 1α. PLoS ONE 2014, 9, e115565, doi:10.1371/journal.pone.0115565.
- Yeh, Y.-M.; Chuang, C.-M.; Chao, K.-C.; Wang, L.-H. MicroRNA-138 Suppresses Ovarian Cancer Cell Invasion and Metastasis by Targeting SOX4 and HIF-1α. J. Cancer 2013, 133, 867–878, doi:10.1002/ijc.28086.
- Hu, Q.; Liu, F.; Yan, T.; Wu, M.; Ye, M.; Shi, G.; Lv, S.; Zhu, X. MicroRNA‑576‑3p Inhibits the Migration and Proangiogenic Abilities of Hypoxia‑treated Glioma Cells Through Hypoxia‑Inducible Factor‑1α. J. Mol. Med. 2019, 43, 2387–2397, doi:10.3892/ijmm.2019.4157.
- He, M.; Zhan, M.; Chen, W.; Xu, S.; Long, M.; Shen, H.; Shi, Y.; Liu, Q.; Mohan, M.; Wang, J. MiR-143-5p Deficiency Triggers EMT and Metastasis by Targeting HIF-1α in Gallbladder Cancer. Physiol. Biochem. 2017, 42, 2078–2092, doi:10.1159/000479903.
- Xue, T.-M.; Tao, L.; Zhang, M.; Zhang, J.; Liu, X.; Chen, G.-F.; Zhu, Y.-J.; Zhang, P.-J. Clinicopathological Significance of MicroRNA-20b Expression in Hepatocellular Carcinoma and Regulation of HIF-1α and VEGF Effect on Cell Biological Behaviour. Markers 2015, 2015, 325176, doi:10.1155/2015/325176.
- Cai, Q.; Wang, Z.; Wang, S.; Weng, M.; Zhou, D.; Li, C.; Wang, J.; Chen, E.; Quan, Z. Long Non-Coding RNA LINC00152 Promotes Gallbladder Cancer Metastasis and Epithelial-Mesenchymal Transition by Regulating HIF-1α via MiR-138. Open Biol. 2017, 7, doi:10.1098/rsob.160247.
- Hong, Q.; Li, O.; Zheng, W.; Xiao, W.-Z.; Zhang, L.; Wu, D.; Cai, G.-Y.; He, J.C.; Chen, X.-M. LncRNA HOTAIR Regulates HIF-1α/AXL Signaling Through Inhibition of MiR-217 in Renal Cell Carcinoma. Cell Death Dis. 2017, 8, e2772, doi:10.1038/cddis.2017.181.
- Tan, H.; Zhao, L. LncRNA Nuclear-Enriched Abundant Transcript 1 Promotes Cell Proliferation and Invasion by Targeting MiR-186-5p/HIF-1α in Osteosarcoma. Cell. Biochem. 2019, 120, 6502–6514, doi:10.1002/jcb.27941.
- Huang, T.; Liu, H.W.; Chen, J.Q.; Wang, S.H.; Hao, L.Q.; Liu, M.; Wang, B. The Long Noncoding RNA PVT1 Functions as a Competing Endogenous RNA by Sponging MiR-186 in Gastric Cancer. Pharmacother. 2017, 88, 302–308, doi:10.1016/j.biopha.2017.01.049.
- Abdel-Haleem, A.M.; Lewis, N.E.; Jamshidi, N.; Mineta, K.; Gao, X.; Gojobori, T. The Emerging Facets of Non-Cancerous Warburg Effect. Endocrinol. 2017, 8, doi:10.3389/fendo.2017.00279.
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. J. Radiat. Biol. 2019, 95, 912–919, doi:10.1080/09553002.2019.1589653.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218, doi:10.1016/j.tibs.2015.12.001.
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree Effects: On the Origin of Cancer Cell Energy Metabolism and of Yeast Glucose Repression. Biophys. Acta BBA Bioenerg. 2011, 1807, 568–576, doi:10.1016/j.bbabio.2010.08.010.
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Endocrinol. 2018, 9, doi:10.3389/fendo.2018.00129.
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Rev. Cancer 2020, 20, 74–88, doi:10.1038/s41568-019-0216-7.
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Genet. 2018, 9, doi:10.3389/fgene.2018.00427.
- Wilkie, M.D.; Anaam, E.A.; Lau, A.S.; Rubbi, C.P.; Jones, T.M.; Boyd, M.T.; Vlatković, N. TP53 Mutations in Head and Neck Cancer Cells Determine the Warburg Phenotypic Switch Creating Metabolic Vulnerabilities and Therapeutic Opportunities for Stratified Therapies. Cancer Lett. 2020, 478, 107–121, doi:10.1016/j.canlet.2020.02.032.
- Varghese, E.; Samuel, S.M.; Líšková, A.; Samec, M.; Kubatka, P.; Büsselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, doi:10.3390/cancers12082252.
- Dabral, S.; Muecke, C.; Valasarajan, C.; Schmoranzer, M.; Wietelmann, A.; Semenza, G.L.; Meister, M.; Muley, T.; Seeger-Nukpezah, T.; Samakovlis, C.; et al. A RASSF1A-HIF1α Loop Drives Warburg Effect in Cancer and Pulmonary Hypertension. Commun. 2019, 10, 2130, doi:10.1038/s41467-019-10044-z.
- Sadlecki, P.; Bodnar, M.; Grabiec, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Zegarska, J.; Walentowicz-Sadlecka, M. The Role of Hypoxia-Inducible Factor-1α, Glucose Transporter-1, (GLUT-1) and Carbon Anhydrase IX in Endometrial Cancer Patients. Available online: https://www.hindawi.com/journals/bmri/2014/616850/ (accessed on 13 October 2020).
- Liu, Y.; Li, Y.; Tian, R.; Liu, W.; Fei, Z.; Long, Q.; Wang, X.; Zhang, X. The Expression and Significance of HIF-1α and GLUT-3 in Glioma. Brain Res. 2009, 1304, 149–154, doi:10.1016/j.brainres.2009.09.083.
- Fujino, M.; Aishima, S.; Shindo, K.; Oda, Y.; Morimatsu, K.; Tsutsumi, K.; Otsuka, T.; Tanaka, M.; Oda, Y. Expression of Glucose Transporter-1 Is Correlated with Hypoxia-Inducible Factor 1 (Alpha) and Malignant Potential in Pancreatic Neuroendocrine Tumors. Lett. 2016, 12, 3337–3344.
- Demaria, M.; Poli, V. PKM2, STAT3 and HIF-1α. JAK STAT 2012, 1, 194–196, doi:10.4161/jkst.20662.
- Dong, G.; Mao, Q.; Xia, W.; Xu, Y.; Wang, J.; Xu, L.; Jiang, F. PKM2 and Cancer: The Function of PKM2 beyond Glycolysis (Review). Lett. 2016, 11, 1980–1986, doi:10.3892/ol.2016.4168.
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnson, D.G.; et al. Pyruvate Kinase M2 Regulates Hif-1α Activity and IL-1β Induction, and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 65–80, doi:10.1016/j.cmet.2014.12.005.
- Tan, V.P.; Miyamoto, S. HK2/Hexokinase-II Integrates Glycolysis and Autophagy to Confer Cellular Protection. Autophagy 2015, 11, 963–964, doi:10.1080/15548627.2015.1042195.
- Nakajima, K.; Kirito, K.; Kawashima, I.; Koshiishi, M.; Kumagai, T.; Mitsumori, T. Aberrant Activation of NF-ΚB and Hypoxia Enhances Glycolytic Enzyme Hexokinase II Expression in B-Cell Lymphoma Through Activation of HIF-1. Blood 2017, 130, 4025–4025, doi:10.1182/blood.V130.Suppl_1.4025.4025.
- Tarrado-Castellarnau, M.; Diaz-Moralli, S.; Polat, I.H.; Sanz-Pamplona, R.; Alenda, C.; Moreno, V.; Castells, A.; Cascante, M. Glyceraldehyde-3-Phosphate Dehydrogenase Is Overexpressed in Colorectal Cancer Onset. Med. Commun. 2017, 2, 6, doi:10.1186/s41231-017-0015-7.
- Liu, K.; Tang, Z.; Huang, A.; Chen, P.; Liu, P.; Yang, J.; Lu, W.; Liao, J.; Sun, Y.; Wen, S.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase Promotes Cancer Growth and Metastasis Through Upregulation of SNAIL Expression. J. Oncol. 2017, 50, 252–262, doi:10.3892/ijo.2016.3774.
- Jadhav, S.; Krynetskaia, N.; Krynetskiy, E. Cellular Response to Anticancer Drugs in Melanoma Cells: Inhibition of Glyceraldehyde 3-Phosphate Dehydrogenase Results in Cell Cycle Arrest and Increased Chemoresistance to Chemotherapeutic Agents. Cancer Res. 2007, 67, 3238–3238.
- Higashimura, Y.; Nakajima, Y.; Yamaji, R.; Harada, N.; Shibasaki, F.; Nakano, Y.; Inui, H. Up-Regulation of Glyceraldehyde-3-Phosphate Dehydrogenase Gene Expression by HIF-1 Activity Depending on Sp1 in Hypoxic Breast Cancer Cells. Biochem. Biophys. 2011, 509, 1–8, doi:10.1016/j.abb.2011.02.011.
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7, doi:10.1016/j.ccell.2018.01.010.
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496, doi:10.3349/ymj.2017.58.3.489.
- Albadari, N.; Deng, S.; Li, W. The Transcriptional Factors HIF-1 and HIF-2 and Their Novel Inhibitors in Cancer Therapy. Expert Opin. Drug Discov. 2019, 14, 667–682, doi:10.1080/17460441.2019.1613370.
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot Trial of EZN-2968, an Antisense Oligonucleotide Inhibitor of Hypoxia-Inducible Factor-1 Alpha (HIF-1α), in Patients with Refractory Solid Tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348, doi:10.1007/s00280-013-2362-z.
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic Targeting of Hypoxia and Hypoxia-Inducible Factors in Cancer. Ther. 2016, 164, 152–169, doi:10.1016/j.pharmthera.2016.04.009.
- Beppu, K.; Nakamura, K.; Linehan, W.M.; Rapisarda, A.; Thiele, C.J. Topotecan Blocks Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor Expression Induced by Insulin-like Growth Factor-I in Neuroblastoma Cells. Cancer Res. 2005, 65, 4775–4781, doi:10.1158/0008-5472.CAN-04-3332.
- Aquino-Gálvez, A.; González-Ávila, G.; Delgado-Tello, J.; Castillejos-López, M.; Mendoza-Milla, C.; Zúñiga, J.; Checa, M.; Maldonado-Martínez, H.A.; Trinidad-López, A.; Cisneros, J.; et al. Effects of 2-Methoxyestradiol on Apoptosis and HIF-1α and HIF-2α Expression in Lung Cancer Cells under Normoxia and Hypoxia. Rep. 2016, 35, 577–583, doi:10.3892/or.2015.4399.
- Narita, T.; Yin, S.; Gelin, C.F.; Moreno, C.S.; Yepes, M.; Nicolaou, K.C.; Van Meir, E.G. Identification of a Novel Small Molecule HIF-1α Translation Inhibitor. Cancer Res. 2009, 15, 6128–6136, doi:10.1158/1078-0432.CCR-08-3180.
- Womeldorff, M.; Gillespie, D.; Jensen, R.L. Hypoxia-Inducible Factor-1 and Associated Upstream and Downstream Proteins in the Pathophysiology and Management of Glioblastoma. Focus 2014, 37, E8, doi:10.3171/2014.9.FOCUS14496.
- Papale, M.; Ferretti, E.; Battaglia, G.; Bellavia, D.; Mai, A.; Tafani, M. EZH2, HIF-1, and Their Inhibitors: An Overview on Pediatric Cancers. Pediatr. 2018, 6, doi:10.3389/fped.2018.00328.
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Oncol. Rep. 2019, 21, 6, doi:10.1007/s11912-019-0752-z.
- Ibrahim, N.O.; Hahn, T.; Franke, C.; Stiehl, D.P.; Wirthner, R.; Wenger, R.H.; Katschinski, D.M. Induction of the Hypoxia-Inducible Factor System by Low Levels of Heat Shock Protein 90 Inhibitors. Cancer Res. 2005, 65, 11094–11100, doi:10.1158/0008-5472.CAN-05-1877.
- Hutt, D.M.; Roth, D.M.; Vignaud, H.; Cullin, C.; Bouchecareilh, M. The Histone Deacetylase Inhibitor, Vorinostat, Represses Hypoxia Inducible Factor 1 Alpha Expression Through Translational Inhibition. PLoS ONE 2014, 9, doi:10.1371/journal.pone.0106224.
- Miranda, E.; Nordgren, I.K.; Male, A.L.; Lawrence, C.E.; Hoakwie, F.; Cuda, F.; Court, W.; Fox, K.R.; Townsend, P.A.; Packham, G.K.; et al. A Cyclic Peptide Inhibitor of HIF-1 Heterodimerization That Inhibits Hypoxia Signaling in Cancer Cells. Am. Chem. Soc. 2013, 135, 10418–10425, doi:10.1021/ja402993u.
- Mangraviti, A.; Raghavan, T.; Volpin, F.; Skuli, N.; Gullotti, D.; Zhou, J.; Asnaghi, L.; Sankey, E.; Liu, A.; Wang, Y.; et al. HIF-1α- Targeting Acriflavine Provides Long Term Survival and Radiological Tumor Response in Brain Cancer Therapy. Rep. 2017, 7, 14978, doi:10.1038/s41598-017-14990-w.
- Lee, K.; Qian, D.Z.; Rey, S.; Wei, H.; Liu, J.O.; Semenza, G.L. Anthracycline Chemotherapy Inhibits HIF-1 Transcriptional Activity and Tumor-Induced Mobilization of Circulating Angiogenic Cells. Natl. Acad. Sci. USA 2009, 106, 2353–2358, doi:10.1073/pnas.0812801106.
- Viziteu, E.; Grandmougin, C.; Goldschmidt, H.; Seckinger, A.; Hose, D.; Klein, B.; Moreaux, J. Chetomin, Targeting HIF-1α/P300 Complex, Exhibits Antitumour Activity in Multiple Myeloma. J. Cancer 2016, 114, 519–523, doi:10.1038/bjc.2016.20.