1. Introduction
Based on their genomic structure, coronaviruses can be divided into four subgroups: α, β, γ, and δ [
50]. SARS-CoV-2 is a β-coronavirus, and similar to other respiratory pathogens, can be spread through droplets generated during talking, sneezing, and coughing. In addition, it can be transmitted by “infected” aerosols generated during medical or dental procedures [
51,
52,
53]. The first step for SARS-CoV-2 infection is the binding to angiotensin-converting enzyme 2 (ACE2), a host cell receptor expressed in lung cells and multiple extrapulmonary tissues [
54,
55]. ACE2 is highly expressed in lung epithelial cells, myocardial cells, the gastrointestinal system, kidney proximal tubule cells, and arterial smooth muscle cells [
56]. Furthermore, the oral mucosa plays a crucial role as a portal for SARS-CoV-2 infection, as oral epithelial cells highly express ACE2 [
57]. Interestingly, ACE2 expression in the nasal epithelium is age-dependent, with lower expression in children, which may explain the higher COVID-19 prevalence in older individuals [
58,
59]. Therefore, although different factors are needed for efficient viral infection, human cells with high ACE2 expression are potentially more susceptible to becoming infected with SARS-CoV-2.
After binding specific receptors, viruses manipulate host cells’ resources in order to replicate. Although SARS-CoV-2 is closely related to another coronavirus, it is considered a new β-coronavirus. To better understand a newly identified virus, many mechanisms can be learned from related strains, including invasion and replication properties [
60]. For instance, SARS-CoV-2 is able to infect and replicate more efficiently in human lung tissues than SARS-CoV [
61]. Some coronaviruses manipulate the mitotic cycle and apoptotic cell death as strategies to promote infection. They can induce cell growth arrest in different cell lines in order to enhance their replication, probably by increasing the availability of deoxynucleotides [
62,
63,
64]. One of the mechanisms used by coronaviruses to induce mitotic arrest is the activation of DNA damage response (DDR) signaling. Xu et al. found that the suppression of ATR (ataxia telangiectasia and Rad3 related), a key modulator of DDR, inhibited viral replication [
63]. Although DDR can act as an antiviral mechanism, some viruses can use DDR factors to stimulate replication [
65]. Consistent with the concept that coronaviruses can induce cell growth arrest to promote their replication, Bouhaddou et al. recently demonstrated that SARS-CoV-2 causes both DNA damage during the early stage of infection and cell cycle arrest [
66]. Therefore, an early cycle inhibitory effect in infected cells may delay their death, and at the same time allow coronaviruses to evade host immune surveillance and facilitate replication [
64].
Following viral infection, cells react and secrete specific chemokines that modulate the recruitment of immune cells; this process represents the first line of defense to reduce virus replication and viral spread [
67,
68]. In contrast, low and dysregulated immune response and reduced levels of antiviral interferons are observed after SARS-CoV-2 infection; however, proinflammatory cytokine secretion remains increased [
69,
70,
71]. High and uncontrolled secretion of proinflammatory cytokines, also known as a “cytokine storm”, can result in severe local tissue injury and also have systemic consequences [
68,
72]. In agreement with this, a cytokine storm caused as a reaction to SARS-CoV-2 infection is a major cause of direct lung injury, acute respiratory distress syndrome (ARDS), multiple organ failure, and unfavorable prognosis [
68,
73]. Therefore, lung tissue damage is the consequence not only of the direct damage from virus, but also from ROS-mediated oxidative stress associated with inflammation [
74].
2. Predictors of Severe Lung COVID-19 Infection
Higher risk of developing severe COVID-19 infection is not the direct consequence of chronological aging itself; it is partially explained by the presence of pre-existing health problems. Indeed, the presence of one or more comorbidities can predict COVID-19 severity in older patients [
75,
76,
77]. Given that severe COVID-19 infection can cause lung damage and dysfunction, it is reasonable to assume that chronic respiratory problems can increase the risk of mortality. Indeed, dyspnea, pneumonia, and COPD are considered the strongest predictive factors for disease severity [
78,
79]. In addition to the role of pre-existing comorbidities, a dysfunctional immune response has an important role in COVID-19 severity. It has been suggested that the combination of age-related decline of the host immune defense (immunosenescence), chronic low-grade systemic inflammation (inflammaging), and SARS-CoV-2 infection can predispose older patients to increased complications [
80]. Along the same line, lymphopenia is also a reliable indicator for disease severity [
81].
CT scans and chest radiographic findings, specific cytokines in serum, and the viral load also help to predict COVID-19 severity and potential complications. Given that bilateral lung involvement and pulmonary opacities increase in frequency during the late stage of infection, they are considered hallmarks of SARS-CoV-2 lung infection [
82,
83,
84]. However, after the onset of symptoms (0–2 days), 56% of patients with SARS-CoV-2 infection had normal chest CT scans. In addition, only 28% had bilateral lung involvement, which suggests a limited predictive value during the early stages of infection [
84]. On the other hand, during lung infection, immune and nonimmune cells display an excessive secretion of proinflammatory cytokines, including interleukin 6 (IL-6) and tumor necrosis factor-α (TNF-α) [
85]. Consistent with this, Del Valle et al. identified that high IL-6 and TNF-α serum levels at the time of hospital admission are strong predictors for disease severity and patient survival [
86]. In this inflammatory setting, the hyperactivation of NF-kB has a crucial role in modulating the secretion of proinflammatory mediators and the immune response [
85]. Another important parameter relevant to predicting disease severity is the viral load, which in patients with severe infection is around 60 times higher than those with mild disease [
87]. Altogether, different parameters can help with the stratification of patients with COVID-19, and the identification of additional clinical factors could increase the predictive capacity of physicians, improving the therapeutic management of patients.
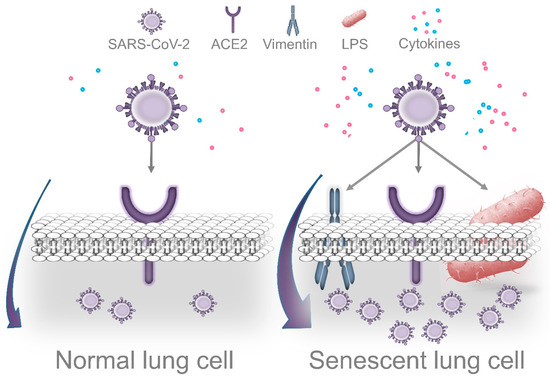
The aspiration of oral fluids or food represents a plausible route for the translocation of oral pathogens into the lungs, especially in geriatric patients. In fact, aspiration pneumonia is one of the most common long-term complications of poor oral hygiene in elderly people, and adequate oral health care can decrease the risk of dying from that condition [
118,
119]. Considering that aspiration pneumonia is often caused by
P. gingivalis [
110,
120], and that oral bacteria have been identified in bronchoalveolar lavage samples from COVID-19 patients [
121], there are at least three interconnected mechanisms that might facilitate SARS-CoV-2 replication in lung cells as a consequence of pre-existing pulmonary infection by periodontal bacteria: first,
P. gingivalis LPS-induced senescence; second, exacerbation of local inflammation in response to SARS-CoV-2; third, decreased immune surveillance caused by periodontal bacteria.