The WNT pathway is one of the major signaling cascades frequently deregulated in human cancer. Binding of WNT ligands to their respective receptors can trigger various downstream signaling cascades centered around cell proliferation, survival or migration. In particular, WNT signaling via the receptor tyrosine kinase-like orphan receptors (RORs) has gained increasing attention in cancer research due to their overexpression in a multitude of tumor entities. In this review, we summarize seminal findings on the structure and expression of the RORs in cancer, their downstream signaling, and its output in regard to tumor cell function.
- ROR1
- ROR2
- cancer
- WNT signaling
1. The ROR Family
The receptor tyrosine kinase-like orphan receptor (ROR) family contains two members: ROR1 and ROR2, which are highly conserved from metazoans to humans. Both belong to the receptor tyrosine kinase (RTK) family, and although they had initially been described as orphan receptors, WNT proteins have meanwhile been identified as their long-missing ligands. ROR1 and ROR2 share an overall amino acid identity of 58% and were already successfully cloned in 1992 from the human neuroblastoma cell line SH-SY5Y [1][12]. The RORs are single-pass transmembrane receptors that harbor an immunoglobulin (Ig)-like domain, a cysteine-rich domain (CRD) and a kringle domain (KRD) in their extracellular part [1] [12] (Figure 1). The CRD of ROR1 and ROR2 is similar to that of the FZD receptors and has been identified as the essential domain for the binding of WNT ligands [2][3][13,14]. RORs are the only RTK family members possessing a KRD, which was shown to be essential for ROR1/ROR2 hetero-oligomerization [4][15].
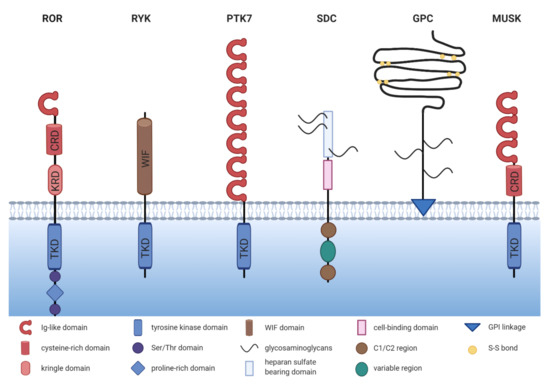
Figure 1. The structure of non-canonical WNT co-receptors. Comparison of the schematic structures of potential non-canonical WNT co-receptors. ROR = receptor tyrosine kinase-like orphan receptor, RYK = receptor tyrosine kinase, PTK7 = protein tyrosine kinase 7, SDC = syndecan, GPC = glypican, MUSK = muscle-specific kinase, TKD = tyrosine kinase domain, KRD = kringle domain, CRD = cysteine-rich domain.
The intracellular domain of the RORs exhibits a tyrosine kinase domain (TKD) and a proline-rich domain (PRD), which is framed by two serine-threonine rich domains [1][12]. The PRD can potentially be recognized by proteins carrying a SRC homology 3 (SH3) domain and thus mediate protein-protein-interactions. Interestingly, the PRDs of ROR1 and ROR2 show relatively low homology, suggesting that the two receptors might differ in their downstream signaling interactors. Within the intracellular domain, ROR1 has 19 tyrosines, which were shown to be phosphorylatable in silico [5][16]. However, the amino acid sequence of the TKD of ROR1 displays six deviations from the canonical tyrosine kinase consensus sequence, amongst which three (C482G, K614R, and L634F) are located in the catalytic center [6][17]. Indeed, the extent of ROR1 autophosphorylation was shown to be negligible [6][7][8][17,18,19]. Likewise, the structure of the TKD of ROR2 shows similarity to RTK-like pseudokinases suggesting a comparable lack of catalytic activity [9][20].
The RORs are predominantly expressed at the plasma membrane, although several reports have claimed a cytoplasmic, and even nuclear location of both [10][11][12][13][14][15][21,22,23,24,25,26]. The different subcellular locations might also derive from the expression of distinct isoforms, which have been described in particular for ROR1 [16][27]. These isoforms comprise the full-length protein expressed on the cell surface (937 aa, 100-105 kDa, unglycosylated, ROR1-001, ENSP00000360120), an intracellular/secreted variant (393 aa, ~44 kDa, ROR1-002, ENSP00000360121), and a third variant of so far unknown localization (388 aa, ~40 kDa ROR1-201, ENSP00000441637). The full-length isoform of ROR1 can be fully N-glycosylated, which results in a 130 kDa protein and permits the cell surface expression of ROR1 [17][28]. In chronic lymphocytic leukemia (CLL) cells a 64 kDa isoform with restricted nuclear expression as well as a 260 kDa isoform were identified, among which the latter probably represents a dimerized ROR1 (homo- or heterodimerized) [18][29]. The 64, 100–105, 130 and 260 kDa isoforms were shown to be phosphorylated at the tyrosine and serine residues and were differentially expressed in CLL patients, depending on their progression status [18][29]. Possible isoforms for ROR2 have not been described yet.
2. Expression of ROR1/2 in Cancer
In order to assess the targetability of ROR1 and ROR2 as novel therapeutic approach, it is important to determine their pattern and level of expression in healthy tissue, in which ROR-targeted therapies might cause undesired off-target effects, and thus toxicity. Furthermore, before choosing which cancer entities might benefit from ROR targeting, it should be noted that the RORs are not uniformly expressed in all cancer tissues, and that their function might differ in the different entities. Therefore, we discuss the current knowledge on the expression of ROR1 and ROR2 in cancer in the following chapter and summarize the findings in Table 1.
Table 1. Expression of ROR1 and ROR2 in cancer.
| Cancer Entity | Expression | 1 | Correlation with Survival |
|---|
| Hematological Malignancies | |||
| acute lymphocytic leukemia | ROR1 + | no correlation found | |
| chronic lymphocytic leukemia | ROR1 + | high ROR1 correlates with poor OS and TFS | |
| diffuse large B cell lymphoma | ROR1 + | n.d. | |
| follicular lymphoma | ROR1 + | n.d. | |
| mantle cell lymphoma | ROR1 + | n.d. | |
| marginal zone lymphoma | ROR1 + | n.d. | |
| multiple myeloma | ROR2 + | n.d. | |
| Solid Tumors | |||
| breast cancer | ROR1 + | high ROR1 correlated with poor OS, MFS and DFS | |
| ROR2 + | high ROR2 correlated with poor DFS | ||
| cervical cancer | ROR2 + | high ROR2 correlated with poor OS and RFS | |
| colorectal cancer | ROR1 + | high ROR1 correlated with poor OS | |
| ROR2 +/− | high ROR2 correlated with poor OS | ||
| endometrial cancer | ROR1 + | high ROR1 correlated with poor OS and PFS | |
| ROR2 + | no correlation found for ROR2 | ||
| gastric cancer | ROR1 + | no correlation found for ROR1 | |
| ROR2 − | n.d. | ||
| glioblastoma | ROR2 + | no correlation found | |
| lung cancer | ROR1 + | high ROR1 correlated with poor OS | |
| ROR2 + | high ROR2 correlated with poor OS | ||
| melanoma | ROR1 + | high ROR1 correlated with poor PRS | |
| mesothelioma | ROR1 + | n.d. | |
| ROR2 + | |||
| ovarian cancer | ROR1 + | high ROR1 correlated with poor OS and DFS | |
| ROR2 +/− | no correlation found for ROR2 | ||
| sarcoma | ROR2 + | high ROR2 correlated with poor OS in GIST high ROR2 correlated with poor DSS in leiomyosarcoma |
|
| pancreatic cancer | ROR1 + | n.d. | |
| ROR2 + | high ROR2 correlated with poor OS | ||
| prostate cancer | ROR2 − | n.d. | |
1 +/− = higher/lower expression compared to normal tissue; n.d. = not determined, OS = overall survival, TFS = therapy-free survival, MFS = metastasis-free survival, DFS = disease-free survival, RFS = recurrence-free survival, PFS = progression-free survival, PRS = post recurrence survival, DSS = disease-specific survival, GIST = gastrointestinal stromal tumor.
2.1. Expression of ROR1/2 in Healthy Tissue
Studies in mice have shown that while ROR1 and ROR2 play an important role in early embryogenesis, their expression is downregulated after birth [1][19][20][21][12,66,67,68]. The expression of ROR1 and ROR2 is not exclusive and can occur in the same cells [20][21][67,68]. In accordance with the mouse studies, in human adult tissues ROR1 was found to be expressed only at very low levels in regions of the esophagus, stomach, duodenum, colon, bladder, uterus, testis, lung, parathyroid, and primary fibroblasts, while expression was higher in pancreatic and adipose tissue [14][22][23][24][25][26][25,30,31,39,69,70]. In the hematologic system, ROR1 was discovered on immature B cells in the bone marrow, and on tonsillar B cells [27][24][28][32,39,71], but not on normal mature B cells, plasma cells, or peripheral blood mononuclear cells (PBMCs) from healthy donors [23][29][24][30][31,33,39,72]. Interestingly, soluble ROR1 has been detected by ELISA in the sera of healthy individuals as well as of patients suffering from CLL, albeit at low concentrations and only in <25% of cases [23][31]. Although the concentrations are likely too low to interfere with ROR1 antibody therapies, this observation suggests that ROR1 might undergo shedding, a finding that warrants further investigation. Among the tissues described as expressing ROR1, the protein was not only located on the cell surface, but was found in the cytoplasm in several cases [22][30]. While different fixation approaches were shown to affect the subcellular localization of ROR1 [11][22], this raises the question, whether the differences result from technical problems, or from the use of distinct ROR1 antibodies directed against the C- or N-terminus recognizing different variants of the protein.
ROR2 mRNA was absent in vital organs, but was weakly detected in the stomach, thyroid as well as in osteoblasts [31][32][33][57,61,73]. Furthermore, the ROR2 protein was detected in healthy colon epithelium by immunoblotting [34][47]. In contrast to ROR1, ROR2 was expressed on normal CD5+ B cells [4][15]. However, a comparative analysis of ROR2 protein expression in adult tissues is still lacking.
2.2. Expression of ROR1/2 in Hematological Malignancies
Already in 2001, gene expression analysis had identified ROR1 as an important component of a signature that distinguishes CLL cells from normal B cells, and other B cell malignancies [35][36][34,35]. The ROR1 gene was found to be 19-fold overexpressed in CLL [35][34]. Although the majority of CLL patients harbor high levels of ROR1-positive tumor cells in blood, ~5% of CLL cases exhibited negligible ROR1 expression [37][36]. ROR1 was described to increase on CLL cells upon disease progression [30][72], and patients with high ROR1 expression had significantly shorter therapy-free survival (TFS) as well as overall survival (OS), indicating that ROR1 is associated with a more aggressive disease [37][36]. ROR1 is not a unique marker for CLL, but is also highly prevalent in other non-Hodgkin lymphoma (NHL) entities, especially in mantle cell lymphoma (MCL) [38][24][37,39]. In addition, 45% of pediatric acute lymphatic leukemia (ALL) patients stained positive for ROR1 in an immunohistochemistry (IHC) study, although no association with survival was detected [22][30].
In contrast to the prominent role of ROR1, initial studies reported that ROR2 was not expressed in hematological malignancies, including CLL [23][30][31,72]. However, this assumption has been challenged as Yu et al. detected ROR2 on freshly isolated CLL cells and demonstrated its hetero-oligomerization with ROR1 [4][15]. Interestingly, ROR2 expression was lost upon longer cultivation of the cells, which might explain the differing observations. Furthermore, in multiple myeloma ROR2 was recently identified by gene expression analysis and IHC to be overexpressed on plasma cells in one third of the investigated patients [39][40], suggesting that further research is warranted to clarify its role in hematological malignancies.
2.3. Expression of ROR1 in Solid Tumors
Overexpression of ROR1 has not only been reported in hematological malignancies, but has also gained increasing attention in solid tumors. IHC analyses have demonstrated a wide-spread expression of ROR1 across many tumor entities with significantly higher expression levels in cancerous than in adjacent normal tissue (Table 1), including a particularly strong expression in melanoma, colon, pancreatic, lung, and breast cancer [11][40][41][22,41,56]. Chang et al. reported that high ROR1 expression was associated with a lower pathological tumor (pT) stage and the absence of perineural invasion in gastric cancer patients who underwent gastrectomy and did not receive neoadjuvant chemotherapy [42][50]. However, so far this remains the only analysis that suggested that ROR1 might be associated with limited disease, while the majority of studies indicated that it is rather associated with aggressiveness and poor survival. ROR1 expression was positively associated with clinical stage and lymph node metastasis in colorectal cancer, for which it served as an independent prognostic marker [43][46]. In ovarian cancer, high ROR1 expression was likewise associated with tumor grade as well as lymph node metastasis [44][58]. In Ewing sarcoma, ROR1 expression was markedly higher in metastases than in localized disease [26][70], and in pancreatic cancer, ROR1 was identified on circulating tumor cells (CTCs) as an essential factor for their invasive phenotype [45][63].
In breast cancer, ROR1 levels seem to vary between the different molecular subtypes, as gene expression data have implied particularly high levels in poorly differentiated and triple-negative breast cancers, the most aggressive breast cancer subtype, in which very high expression (top 10%) was associated with shorter OS [40][41]. Similar results were obtained from an IHC study in which 57% of triple-negative breast cancers stained highly positive for ROR1, whereas signals were detected in only 12% of the estrogen receptor (ER) and progesterone receptor (PR)-positive patients, and no expression at all was found in Her2/Neu-positive breast cancers [14][25]. Interestingly, breast cancer brain metastases have likewise been shown to highly express ROR1 [46][74], hinting at its role in metastatic spread.
ROR1 has also been extensively studied in lung cancer. Since it was identified as a transcriptional target of homeobox protein Nkx-2.1 [47][75], a major lineage-survival oncogene in lung cancer, it is not surprising that many lung tumors harbor ROR1. Comparable to breast cancer, ROR1 was expressed in certain subtypes and was particularly high in lung adenocarcinoma (40–65% positive), while squamous carcinomas stained positive at much lower frequencies [11][14][48][22,25,76]. In lung adenocarcinoma, ROR1 functions as an independent prognostic predictor for OS [31][57]. Both ROR1 and ROR2 were found to be highly expressed in a lung cancer cell line derived from CTCs from a patient with relapsed small cell lung cancer [49][77]. This fits to the observation in two patients with ROR1-negative primary tumors, in whom the metastases gained ROR1 [14][25]. While these observations suggest that ROR1 might be involved in metastasis, the same study also showed that 40% of ROR1-positive primary tumors lost their ROR1 expression in the matched metastatic lesions [14][25], thus questioning this concept. Taken together, this raises the question whether ROR1 solely functions as a bystander molecule upregulated during tumor progression, or whether the functional consequence of WNT/ROR signaling is highly context-specific as discussed later.
2.4. Expression of ROR2 in Solid Tumors—It Is Not Always That Simple
Compared with ROR1, the expression profile of ROR2 in cancer is much more heterogeneous (Table 1). Although most studies describe ROR2 as a positive prognostic factor in a variety of solid tumor entities, a limited number of reports observed a loss of ROR2 in cancerous compared to normal tissue and an association of high ROR2 expression with a favorable outcome. One problem that might have caused some of the contradictory results is that several commercially available antibodies have been reported to display non-specific binding [49][77], thus bringing into question the reliability of the IHC results obtained with these antibodies.
A particularly high percentage of ROR2-positive tumors was found in breast cancer (87%), glioblastoma (>90%), and neuroblastoma (80%) [13][50][51][24,53,78]. Interestingly, ROR2 was expressed in only 20 of 48 of primary melanoma cases, while all of the investigated visceral or subcutaneous metastases (48/48) stained positive [52][79]. Similar observations have been reported for lymph node and brain metastases in breast cancer [46][53][74,80]. Indeed, ROR2 expression was found to correlate with tumor stage and/or lymph node metastasis in lung [54][55], cervical [55][45], and breast cancer [13][24]. Taken together, these studies point to a specific role for ROR2 in invasive growth and metastasis of the named tumor entities.
In contrast, downregulation of ROR2 in solid tumor cells, compared with adjacent normal tissue, has been described for gastric [56] [52] and prostate carcinoma [57][65].
Contradictory findings for ROR2 have been reported for endometrial, colorectal, and ovarian cancer. Initially, the study by Lara et al. demonstrated promoter hypermethylation and consequently repressed expression of ROR2 in 5/8 colorectal cancer cell lines and 3/6 patients [34][47]. In contrast, another study using quantitative real-time PCR detected a significantly higher expression of ROR2 in colorectal cancer compared with adjacent normal tissue [12][23]. In ovarian cancer, a comparative analysis has revealed that ROR1 was largely expressed in cancer cells, while the number of patients with ROR2-positive tumors was significantly lower [58][81]. While ROR2 was found upregulated in a large ovarian carcinoma cohort compared with normal ovarian tissue [59], another report investigating aggressive high-grade serous ovarian carcinoma has claimed the opposite [60]. Whether these controversial findings reflect context-specific functions of ROR2, or whether they arise from non-specific antibody binding in IHC analyses remains to be clarified.
2.5. ROR1/2 in the Tumor Stroma
While research has traditionally focused on the expression and function of the two RORs in cancer cells, several recent reports support the notion that they also play a relevant role in the cancer-associated microenvironment. In ovarian cancer, stroma cell expression of ROR1 was lower than in the tumor cells, while the opposite was true for ROR2 [58][81]. Interestingly, the expression levels of ROR2 increased even further in metastatic tissue compared with matched primary tumor samples [58][81]. In pancreatic cancer stromal ROR2 correlated with regional lymph node invasion and represented an independent prognostic factor [61][64]. While the significance of stromal ROR2 remains elusive so far, novel data have shown that ROR2 is upregulated in reactive astrocytes at brain injury sites [62][82]. Concordantly, both ROR1 and ROR2 were induced in skeletal muscles by pro-inflammatory cytokines (e.g., TNF-α, IL-1β) released after injury [63][83]. These findings suggest that the upregulation of the RORs might occur in stromal cells due to the inflammatory conditions often observed in the tumor microenvironment.
3. The Function of ROR1/2 in Cancer
WNT signaling is known to regulate many key cellular processes, and aberrant regulation of this pathway has been found to contribute to the development and progression of many human cancers. Likewise, the deregulated expression of the WNT co-receptors ROR1 and ROR2 has been associated with several cellular features that promote malignancy, namely cell proliferation, survival, migration/invasion, and stemness, as it will be discussed in the following chapters.
While most studies identified ROR1 as an oncogene, the role of ROR2 in cancer is less clear and has been controversially discussed. Studies in osteosarcoma, melanoma, CLL, breast, and renal cancer have claimed that ROR2 acts as an oncogene in these entities. In contrast, in prostate, colorectal and endometrial cancer some studies attribute a tumor suppressive function to ROR2. Whether the output of ROR2 signaling differs especially in colorectal and endometrial cancer, two entities substantially driven by the β-catenin-dependent, canonical WNT pathway, or whether different organ microenvironments with varying repertoires of FZDs and alternative WNT co-receptors are responsible for the diverging effects is not yet fully understood.
3.1. ROR1/2 in Cell Proliferation and Survival
More than a decade ago both ROR1 and ROR2 were identified as pro-survival kinases by an RNAi screen in HeLa cells [64][84]. This finding spiked the interest of cancer biologists early on to not only evaluate the potential of these novel receptors as cancer biomarkers, but also to consider their functional involvement in tumor development and progression. Meanwhile it became evident that ROR1 plays a major role in cancer cell survival by promoting proliferation, while at the same time counteracting apoptosis. This has not only been observed in a multitude of in vitro studies on cancer cell lines [4][11][18][40][65][66][41][31][47][48][15,22,29,41,48,51,56,57,75,76], but was also confirmed in xenograft studies in vivo [40][47][41,75]. In a mouse model for human CLL, the B cell-restricted expression of ROR1 accelerated leukemogenesis through an interaction of ROR1 with the T cell leukemia 1 (TCL1) oncogene, which activated pro-survival signaling via the protein kinase AKT and resulted in leukemia cell proliferation and resistance to apoptosis [67][85]. In line with this, transcriptomic analyses of ROR1-expressing patient-derived CLL cells revealed a gene expression signature associated with protein kinase activation, AKT-related signaling factors and proliferation, while ROR1-negative CLL cells were characterized by subnetworks associated with apoptosis and consequential RNA processing and decay [37][36]. Interestingly, recent observations indicate that the pro-survival function of ROR1 not only strongly supports leukemogenesis, but is also linked with the development of leukemia. In mice, the knockout of miRNA miR-15/16, leading to the overexpression of the pro-survival factor BCL2 as well as ROR1, induced the development of B cell lymphoma in 23% of the animals, while 77% developed an aggressive acute myeloid leukemia (AML) [68][86]. This finding indicates a great need for further investigation of ROR1, not only in lymphoma, but also in AML, and suggests that combined targeting of ROR1 (e.g., cirmtuzumab) and BCL2 (e.g., venetoclax) might be worth considering. Taken together, these observations support the notion that the pro-survival function of ROR1 is also valid in vivo.
Mechanistic studies have shown that the effect of ROR1 on proliferation and apoptosis required phosphorylation of its PRD [5][16], thus suggesting an activation of ROR1-dependent downstream signaling. Concordantly, additional stimulation of ROR1 with WNT5A supported its effect on cell survival in CLL cells [29][37][33,36]. Blockade of ROR1 in lung cancer cells was found to suppress expression of CDK4 and CCNE1, two important cell cycle regulators, as well as Bcl-XL and Bcl-2, two critical anti-apoptotic factors, while it increased the expression of several pro-apoptotic factors including Bak, caspase-3, and caspase-7 [69][87]. However, whether these factors are direct target genes of ROR1 and which signaling pathways are implemented in their upstream regulation remains elusive so far. Moreover, two novel mechanisms that might contribute to the pro-survival function of ROR1 have recently been recognized: on the one hand, ROR1 inhibition was described to increase p53 activity [70][88], on the other hand, an involvement of ROR1 in autophagy was suggested [69][87]. Further research is required to understand the role of ROR1 in these processes in general as well as in the context of cancer.
In ovarian cancer ROR1 was described to have a synergistic effect with ROR2 on cell proliferation, since only a double knockdown led to a significant reduction in tumor cell proliferation [59]. Similar to ROR1, ROR2 supported proliferation and tumor growth, while inhibiting apoptosis both in vitro as well as in vivo in mouse models of osteosarcoma, breast and renal cancer [71][72][73][74][44,89,90,91]. Likewise, the expression of several cell cycle-related genes including CDK1/2/4, CCNE1, CCND1/2, PCNA, and MKI67 has been described to be regulated by ROR2 in these cancer entities [53][73][75][80,90,92]. In contrast, in a study on ovarian cancer the induction of ROR2 expression in the tumor cells led to endoplasmic reticulum stress and decreased cell viability [60].
A study in NIH3T3 fibroblasts has demonstrated that ROR2 was dynamically regulated during the cell cycle, probably because it is under the transcriptional control of the cell cycle regulator E2F1 [76][93]. ROR2 knockdown led to an accumulation of osteosarcoma cells in the G0/G1 phase [75] [92] suggesting that ROR2 itself is essential for cell cycle progression. Downregulation of ROR2 not only decreased the expression of various E2F1 target genes, but also induced Forkhead box protein O1 (FoxO) target genes [76][93], thus favoring apoptosis induction.
Recent studies have implied that ROR1 and ROR2 might not only promote cell survival through the regulation of pro-mitotic factors, but also by controlling cellular structures implicated in pro-survival signaling, such as filopodia [17][77][28,94]. In gastric cancer cells, ROR2 was described as stimulating the number and length of specific signaling filopodia, so-called cytonemes, which transport WNT ligands to neighboring cells, and thus activate canonical WNT signaling responses stimulating cell proliferation [78][95]. ROR1, in contrast, was found to interact with Caveolin-1, Cavin-1, and Cavin-3 and was not only required for correct caveolae formation, but also for caveolae-dependent endocytosis and induction of signaling responses [79][80][96,97]. As several pro-survival RTKs (e.g., epidermal growth factor receptor (EGFR), hepatocyte growth factor receptor (MET), Insulin-like growth factor 1 receptor (IGF1R)) depend on these functions, inhibiting ROR1 in lung cancer was proposed as a novel shortcut to bypass resistance to EGFR tyrosine kinase inhibitors [79][96]. Indeed, targeting ROR1 helped to reinstall sensitivity to erlotinib treatment in resistant lung cancer cell lines [81][98], thus further underlining its potential as a therapeutic target.
3.2. ROR1/2 in Therapy Resistance and Cancer Stem Cells
Next to the established function of the RORs in cell proliferation, another detrimental consequence of active WNT/ROR signaling in cancer has currently emerged, namely the establishment of resistant tumor cell clones. Several studies have reported the upregulation of ROR1 or ROR2 in chemoresistant cancer cell lines [70][82] [88,99] as well as in patient-derived tissues after chemo- or targeted therapy [83][84][85][100,101,102]. In melanoma, O’Connell et al. initially observed opposing roles for ROR1 and ROR2: melanoma cell lines resistant to inhibition of the serine/threonine-protein kinase B-raf (BRAF) were characterized by upregulated ROR2, but downregulated ROR1 [86][103]. Another study on uveal melanoma cell lines described upregulation of both RORs after mitogen-activated protein kinase kinase (MEK) inhibition and implicated them in the induction of a pro-survival AKT signaling response [87][104]. In ALL cell line models, and primary cells, the knockdown of ROR1 enhanced sensitivity to several small molecule inhibitors in current clinical use [88][105]. Addressing the question of how ROR1/2 can mediate therapy resistance, three different modes are currently being discussed:
Firstly, WNT/ROR1 signaling was identified as a rescue pathway that can induce nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT), and MEK/extracellular-signal-regulated kinase (ERK) activation independent of B cell receptor (BCR)/Bruton tyrosine kinase (BTK) in CLL and MCL [7][89][90][18,106,107]. This activation was mediated via the formation of a complex between ROR1 and CD19 [90] [107] and represented a novel resistance mechanism for treatment strategies centered around BTK/BCR inhibition. Indeed, synergistic effects have been shown for co-targeting both ROR1 and the BCR or BCL-2 family, underlining the large potential for ROR1-targeted therapies in overcoming MCL and CLL drug resistance [89][91][106,108].
Secondly, ROR1 was shown to upregulate the expression of ATP-dependent translocase ABCB1, a multi-drug efflux pump, and thus facilitate drug export from cancer cells [70][88]. Indeed, targeting ROR1 in ovarian cancer increased the efficacy of second mitochondria-derived activator of caspase (SMAC) mimetics and taxanes [92][109]. In line, combining paclitaxel treatment with ROR1 blockade significantly enhanced tumor growth inhibition in a patient-derived xenograft (PDX) model for breast cancer [85][102].
Thirdly, ROR1 has been associated with treatment-resistant cancer stem cells as reviewed in [93][110]. This conclusion was based on observations in ovarian cancer stem cells, in which ROR1 was highly expressed and regulated the expression of the self-renewal marker polycomb complex protein BMI-1 [94][111]. Meanwhile, studies on breast cancer primary cells have likewise identified a population of cells carrying the cancer stem cell markers ALDH1+/CD44+/CD24low that displayed particularly high ROR1 expression and showed enhanced growth of tumor spheres in vitro as well as tumorigenicity in vivo [83][85][100,102]. These ROR1+ cells were characterized by enhanced stemness frequency and expressed markers typically associated with stem cells such as e.g., POU5F1, NANOG, or SOX2 [83][100]. Concordantly, in a phase 1 trial on cirmtuzumab, a monoclonal antibody directed against ROR1, gene expression signatures associated with stemness were markedly lower in post-treatment samples of CLL patients [95][112]. Taken together, these observations suggest that ROR1 is indeed expressed on cancer stem cells and might thus be involved in therapy resistance and relapse. Whether ROR2 fulfills the same functions as ROR1 in the development of drug resistance or whether it acts via distinct mechanisms remains to be elucidated.
3.3. ROR1/2 in EMT and Metastasis
The clinical observations that ROR1 and ROR2 are associated with disease progression and metastasis in many cancer patients have inspired researchers to investigate the role of both co-receptors in the underlying cellular processes. In CLL, stimulation of ROR1-expressing cancer cells with WNT5A stimulated their migratory potential and CXCL12/CCL19-directed chemotaxis, while this effect was not observed in ROR1-negative cells [29][37][33,36]. Similar observations have been made in breast cancer in which ROR1 knockdown decreased CXCR4 expression resulting in decreased chemotaxis of the cells towards a CXCL12 gradient [96][42]. Moreover, in several in vitro solid tumor models ROR1 knockdown reduced cellular migration/invasion. These include Ewing sarcoma [26][70], glioblastoma spheres [97][113], mesothelioma [31][57], breast cancer [96][98][42,114], ovarian cancer [59] [59] or melanoma [41][56]. Another report on melanoma observed that ROR1 was associated with a poorly invasive phenotype, and its knockdown increased invasion in vitro as well as metastasis formation in vivo. However, these diverging results might be caused by the simultaneous upregulation of compensatory ROR2/WNT5A [86][103]. In line with cell motility, ROR1 was found to regulate cell adhesion [41][56].
First reports about a pro-invasive function of ROR2 stem from studies on HeLa cells showing that WNT5A-ROR2 mediated polarized cell migration in this cell line [99][115]. This has since been confirmed in several solid tumor entities in which knockdown of ROR2 resulted in decreased migration and/or invasion, e.g., in mesothelioma [31][57], melanoma [52][79], renal cancer [72][89], breast cancer [98][114], ovarian cancer [59][82][100][101][59,99,116,117], prostate cancer [102][118], leiomyosarcoma, gastrointestinal stroma tumors [103][62], and osteosarcoma [32][61]. Taken together, the data imply that both ROR1 and ROR2 are associated with highly malignant cancer cell phenotypes characterized by high motility and aggressiveness.
The pro-migratory and -invasive functions of ROR1 and ROR2 might not only result from the activation of a typical non-canonical WNT signaling response, which will be discussed in the next chapter, but have been associated with the induction of an epithelial-to-mesenchymal-transition (EMT) phenotype. EMT is a reversible cellular program in which cancer cells lose their adhesive, epithelial characteristics and gain a more motile, mesenchymal phenotype, which drives tumor spreading. ROR1-high compared to ROR1-low breast cancer tumors initially showed high expression of genes typically associated with mesenchymal cells [96][42]. In renal cell carcinoma patients expression of ROR2 was found to be tightly associated with the EMT regulator TWIST and the matrix-degrading enzyme MMP2 [72][89]. Further in vitro studies have indeed confirmed higher expression of typical epithelial factors (e.g., CK19, CDH1) and lower expression of mesenchymal factors (e.g., SNAI2, ZEB1, VIM) after ROR1 or ROR2 knockdown in cancer cells. This is indicative of a role for both co-receptors in fostering EMT [96][41][53][94][97][101][42,56,80,111,113,117]. One explanation why the RORs are so closely associated with the EMT program is that their expression has been revealed to be under the control of key EMT-inducing transcription factors. For instance, TWIST was demonstrated to activate the transcription of the ROR1 gene [104][119], while the expression of ROR2 was found to be regulated by SNAI1 [105][120]. Another report claimed that ROR2 itself was responsible for regulating SNAI1 expression [101][117], raising the chicken and egg question—which comes first?
The set of EMT genes regulated by ROR2, in particular, comprises the matrix metalloproteinases MMP2 and MMP9 [53][72][105][80,89,120]. As these enzymes are critical for degrading the extracellular matrix, their enhanced expression in ROR-positive cells might explain their large potential for invasive growth. Another mechanism by which especially ROR2 promotes invasiveness is the induction of invadopodia formation [77][106][94,121], i.e., special plasma membrane protrusions that can serve as specific sites for the secretion of matrix-degrading enzymes. Recently, ROR2 was found to upregulate the expression of intraflagellar transport 20 (IFT20), a protein originally involved in the microtubule-based transport in the cilium. IFT20 regulates Golgi structure and function which are required for correct cell polarization and subsequent invasive behavior [107][122]. WNT5A-ROR was also described as inducing the degradation of the kinesin KIF26b which was identified as a novel cytoskeletal effector of WNT/ROR signaling and regulated cell migration. Although the mechanism of WNT/ROR/KIF26b signaling is not fully understood yet, it was proposed that it fine-tunes de-adhesion and/or retraction at the trailing edge of migrating cells [108][123]. Thus, it seems that the RORs are substantially involved in regulating processes associated with cytoskeleton dynamics that finally result in increased motility and invasiveness.
Evidence suggests that these findings are also valid in vivo, as ROR1 knockdown significantly reduced the capacity of breast cancer cells intravenously injected into immunodeficient mice to invade the lung 24 to 72 h after injection [96][42], pointing to a role for ROR1 in extravasation and adhesion due to its control of the EMT phenotype. Similar observations have been made for ROR2: multiple myeloma cells with stable ROR2 knockdown were mostly unable to home into the bone marrow due to a defective adhesion to the bone marrow microenvironment [39][40]. Consequently, cancer cells with reduced expression of ROR1 or ROR2 were characterized by a significantly impaired potential in forming metastases in mouse models of melanoma or breast cancer [96][52][42,79].
However, some reports claim that in some cancer subtypes, the RORs are rather associated with less motile and less invasive tumor cells. For instance, when discussing ROR1 and EMT, the special case of hepatocellular carcinoma should be mentioned, since in that tumor entity ROR1 was associated more with an epithelial rather than a mesenchymal phenotype indicating that it might play an opposing role here [109][124]. For ROR2, especially in colorectal and prostate cancer conflicting data have been obtained. For the latter, ROR2 knockdown was initially found to reduce cell invasion [102][118]. However, a recent study reported a reduction in tumor motility and invasiveness upon ROR2 overexpression [57][65], again raising the question whether the regulation of ROR expression levels is critical for determining their functional impact. Moreover, WNT5A secreted by the osteoblastic niche was observed to induce dormancy in prostate cancer cells via ROR2 by repressing canonical WNT signaling and thus inhibiting bone metastasis [110][125]. Whether this interesting finding is specific for prostate cancer, or for bone metastasis, or if it also occurs in other tumor entities remains unknown. In the second example of colorectal carcinoma, hypermethylation, and thus silencing, of the ROR2 promoter was identified as an early event in carcinogenesis. Decreased ROR2 levels were associated with increased proliferation and migration, but with greatly reduced invasiveness [34][111][47,126]. While the early downregulation of ROR2 might represent a way to circumvent its inhibitory influence on canonical WNT signaling, the observed differential regulation of migration and invasion could indicate that ROR2 is indeed responsible for dynamically fine-tuning these cellular processes. In general, ROR2 seems to have tumor suppressive effects particularly in tumors driven by canonical WNT signaling (e.g., colorectal, prostate, or endometrial cancer), whereas it acts rather as an oncogene in tumors with predominantly active non-canonical WNT signaling (e.g., melanoma, breast cancer). This suggests that the different signaling contexts have a significant impact on the functional output of WNT/ROR signaling.