Multiple myeloma (MM) is an incurable plasma cell malignancy characterized by genomic instability. MM cells present various forms of genetic instability, including chromosomal instability, microsatellite instability, and base-pair alterations, as well as changes in chromosome number. The tumor microenvironment and an abnormal DNA repair function affect genetic instability in this disease. In addition, states of the tumor microenvironment itself, such as inflammation and hypoxia, influence the DNA damage response, which includes DNA repair mechanisms, cell cycle checkpoints, and apoptotic pathways. Unrepaired DNA damage in tumor cells has been shown to exacerbate genomic instability and aberrant features that enable MM progression and drug resistance.
- multiple myeloma
- DNA repair
- genomic instability
- DNA damage response
- base excision repair
- homologous recombination
1. Introduction
Multiple myeloma (MM), which accounts for 1% of all cancers and approximately 10% of all hematologic malignancies, is characterized by the clonal proliferation of plasma cells [1][2]. The development and introduction of therapeutics, such as the immunomodulatory drugs thalidomide and lenalidomide and the proteasome inhibitor (PI) bortezomib, have led to improved prognosis of MM [1]. Before the introduction of alkylating agents in MM treatment, the median survival time of symptomatic patients was <1 year, and the introduction of melphalan in the 1960s resulted in improved survival [3][4]. High-dose melphalan (HDM) followed by autologous stem cell transplantation (ASCT) has become a standard of care for younger patients after bortezomib-based induction regimens [5][6]. In addition, the introduction of novel agents, particularly bortezomib combined with lenalidomide plus dexamethasone, has improved the outcome of patients who are ineligible for ASCT [7]. Recently, carfilzomib [8], pomalidomide [9], panobinostat [10], ixazomib [11], elotuzumab [12], daratumumab [13], isatuximab [14], and selinexor [15] have been approved by the Food and Drug Administration (FDA) for the treatment of relapsed MM and promise to improve outcomes. Currently, numerous combination therapies are available and include immunomodulatory drugs, PIs, histone deacetylase inhibitors, and monoclonal antibodies. However, MM remains an incurable disease, and new therapeutic strategies are still needed [1].
Multiple myeloma (MM), which accounts for 1% of all cancers and approximately 10% of all hematologic malignancies, is characterized by the clonal proliferation of plasma cells [1,2]. The development and introduction of therapeutics, such as the immunomodulatory drugs thalidomide and lenalidomide and the proteasome inhibitor (PI) bortezomib, have led to improved prognosis of MM [1]. Before the introduction of alkylating agents in MM treatment, the median survival time of symptomatic patients was <1 year, and the introduction of melphalan in the 1960s resulted in improved survival [3,4]. High-dose melphalan (HDM) followed by autologous stem cell transplantation (ASCT) has become a standard of care for younger patients after bortezomib-based induction regimens [5,6]. In addition, the introduction of novel agents, particularly bortezomib combined with lenalidomide plus dexamethasone, has improved the outcome of patients who are ineligible for ASCT [7]. Recently, carfilzomib [8], pomalidomide [9], panobinostat [10], ixazomib [11], elotuzumab [12], daratumumab [13], isatuximab [14], and selinexor [15] have been approved by the Food and Drug Administration (FDA) for the treatment of relapsed MM and promise to improve outcomes. Currently, numerous combination therapies are available and include immunomodulatory drugs, PIs, histone deacetylase inhibitors, and monoclonal antibodies. However, MM remains an incurable disease, and new therapeutic strategies are still needed [1].
MM cells present genomic instability [16], whose molecular basis is not fully understood. Recently, it has been reported that the DNA damage response (DDR) may influence genomic changes in MM [17][18][19][20]. An abnormal DNA repair function may provide an alternative explanation for aneuploidy and chromosomal rearrangements. Furthermore, the tumor microenvironment may be mutagenic and constitute a significant source of genetic instability, affecting genomic stability and tumor resistance to treatment (
MM cells present genomic instability [16], whose molecular basis is not fully understood. Recently, it has been reported that the DNA damage response (DDR) may influence genomic changes in MM [17,18,19,20]. An abnormal DNA repair function may provide an alternative explanation for aneuploidy and chromosomal rearrangements. Furthermore, the tumor microenvironment may be mutagenic and constitute a significant source of genetic instability, affecting genomic stability and tumor resistance to treatment (
Figure 1) [21][22][23]. This review provides an overview of the DDR, with a special focus on its function on MM. We will also discuss the role of DNA repair in regulating the metabolism and progression of MM cells.
) [21,22,23]. This review provides an overview of the DDR, with a special focus on its function on MM. We will also discuss the role of DNA repair in regulating the metabolism and progression of MM cells.
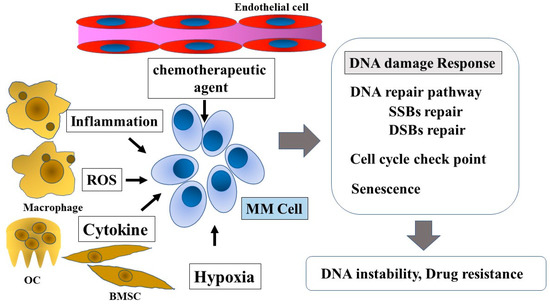
Figure 1.
DNA damage response in multiple myeloma and the tumor microenvironment. Tumor microenvironment factors, such as chemotherapeutic agents, inflammation, ROS, cytokines, genotoxic stress, and hypoxia, influence the DNA damage response in MM cells. The DNA damage response affects disease progression and drug resistance. ROS, reactive oxygen species; OC, osteoclast; BMSC, bone marrow stromal cell; MM Cell, multiple myeloma cell; SSBs, single-strand breaks; DSBs, double-strand breaks.
2. Genomic Instability in MM
Most cancers are characterized by genomic instability [24][25][26], as cells often display chromosomal translocation, aneuploidy, and unchecked cell proliferation. Genomic instability is also a hallmark of MM cells, manifesting largely as whole chromosome- or translocation-based aneuploidy [22][27]. MM cells present various forms of genomic instability, including chromosomal instability (CIN), microsatellite instability, and increased frequency of mutations [28]. Especially, copy number and structural changes due to CIN are common features of MM. Numerical CIN is associated with copy number alterations (CNAs) of entire chromosomes or amplifications and deletions of chromosome arms [29]. The MM genome is also characterized by aberrations caused by structural CIN, such as chromosomal rearrangements, inversions, or complex reassembly of different parts of the chromosomes [29]. Centrosomes play important roles in processes that ensure proper segregation of chromosomes in human cells [30]. Chromosome abnormalities may be associated with centrosome amplification or alterations in the spindle assembly checkpoint. A high centrosome index—prompted by an abnormally high expression of genes encoding the main centrosome proteins—showed an independent prognostic factor in MM [31]. Moreover, dysregulations of the cell-cycle also affect CIN, the most prominent example of which is dysregulation of cyclin D expression—present in both hyperdiploid and non-hyperdiploid MM [28][32]. Furthermore, CIN is affected by the tumor microenvironment, such as hypoxia [33]. Depending on the genes affected, CIN can contribute to a further increase in genome instability [28].
Most cancers are characterized by genomic instability [27,28,29], as cells often display chromosomal translocation, aneuploidy, and unchecked cell proliferation. Genomic instability is also a hallmark of MM cells, manifesting largely as whole chromosome- or translocation-based aneuploidy [22,30]. MM cells present various forms of genomic instability, including chromosomal instability (CIN), microsatellite instability, and increased frequency of mutations [31]. Especially, copy number and structural changes due to CIN are common features of MM. Numerical CIN is associated with copy number alterations (CNAs) of entire chromosomes or amplifications and deletions of chromosome arms [32]. The MM genome is also characterized by aberrations caused by structural CIN, such as chromosomal rearrangements, inversions, or complex reassembly of different parts of the chromosomes [32]. Centrosomes play important roles in processes that ensure proper segregation of chromosomes in human cells [33]. Chromosome abnormalities may be associated with centrosome amplification or alterations in the spindle assembly checkpoint. A high centrosome index—prompted by an abnormally high expression of genes encoding the main centrosome proteins—showed an independent prognostic factor in MM [34]. Moreover, dysregulations of the cell-cycle also affect CIN, the most prominent example of which is dysregulation of cyclin D expression—present in both hyperdiploid and non-hyperdiploid MM [31,35]. Furthermore, CIN is affected by the tumor microenvironment, such as hypoxia [36]. Depending on the genes affected, CIN can contribute to a further increase in genome instability [31].
MM patients are broadly grouped into hyperdiploid or non-hyperdiploid groups, depending on the number of chromosomes present in the MM cells. Hyperdiploid tumors are characterized by trisomies of one or more of the odd-numbered chromosomes 3, 7, 9, 11, 15, or 17, while the majority of non-hyperdiploid tumors display a translocation involving the
IGH
locus on chromosome 14 and one of the five recurrent translocation partners on chromosomes 4, 6, 11, 16, and 20. Five chromosomal partners account for the majority of primary
IGH
translocations: 11q13 (
CCND1
), 6p21 (
CCND3
), 4p16 (
FGFR3
and
NSD2
), and 16q23 (
MAF) [34][35]. These events are present in most patients with monoclonal gammopathy of undetermined significance (MGUS), and secondary genetic alterations occur with an increased incidence in disease progression from MGUS to MM [36]. These secondary events include translocations, deletions, and chromosome gains, involving genes such as
) [37,38]. These events are present in most patients with monoclonal gammopathy of undetermined significance (MGUS), and secondary genetic alterations occur with an increased incidence in disease progression from MGUS to MM [39]. These secondary events include translocations, deletions, and chromosome gains, involving genes such as
MYC
,
KRAS
,
NRAS
, and
TP53, which are involved in the DDR. Secondary events are detected in the late stage of the disease and affect its progression [36][37]. Among these alterations, t (4:14) and del(17p) were associated with poor outcomes [38]. The complexity of the genomic alteration characteristics is correlated with different grades of CIN. These observations strongly implicate CIN as an important biological and prognostic marker in MM [28][39].
, which are involved in the DDR. Secondary events are detected in the late stage of the disease and affect its progression [39,40]. Among these alterations, t (4:14) and del(17p) were associated with poor outcomes [41]. The complexity of the genomic alteration characteristics is correlated with different grades of CIN. These observations strongly implicate CIN as an important biological and prognostic marker in MM [31,42].
Current treatment modalities of MM include alkylating agents, PIs, and anthracyclines, all of which induce excessive DNA damage [23]. DNA-damaging agents, including chemotherapy, can induce mutations and destabilize the genome, resulting in secondary malignancies or allowing the selection of resistant cancer cell clones [23][40]. Melphalan is a nitrogen mustard known to induce mono-alkylation of adenine and guanine, and interstrand DNA crosslinks (ICLs) involving guanine. The number of melphalan-induced ICLs is correlated with its concentration [41]. Cyclophosphamide is also an alkylating agent that induces ICLs. In a cohort of patients with MM treated with HDM and ASCT, polymorphisms in DNA repair genes, including poly(ADP-ribose) polymerase1 (
Current treatment modalities of MM include alkylating agents, PIs, and anthracyclines, all of which induce excessive DNA damage [23]. DNA-damaging agents, including chemotherapy, can induce mutations and destabilize the genome, resulting in secondary malignancies or allowing the selection of resistant cancer cell clones [23,24]. Melphalan is a nitrogen mustard known to induce mono-alkylation of adenine and guanine, and interstrand DNA crosslinks (ICLs) involving guanine. The number of melphalan-induced ICLs is correlated with its concentration [43]. Cyclophosphamide is also an alkylating agent that induces ICLs. In a cohort of patients with MM treated with HDM and ASCT, polymorphisms in DNA repair genes, including poly(ADP-ribose) polymerase1 (
PARP1
),
RAD51
,
PCNA
,
OGG1
,
XPC
,
BRCA1
,
ERCC1
,
BARD1
, and
TP53BP1, were associated with the outcome and overall survival [42]. These genes are significantly enriched in genes involved in homologous recombination repair (HRR) and nucleotide excision repair (NER), necessary for ICL repair. Bortezomib is the first PI approved by the FDA for the treatment of newly diagnosed and relapsed/refractory MM. This drug can bind to and form a complex with the active site of the threonine hydroxyl group in the β5 subunit of the proteasome and block its chymotrypsin-like activity [43]. Recent studies on proteasome inhibition in MM cells have revealed that the accumulation of unfolded proteins in the endoplasmic reticulum, the so-called endoplasmic reticulum stress, triggers that of several pro-apoptotic factors and cell stressors, such as reactive oxygen species (ROS). PI affects DNA repair via depletion of the free ubiquitin pool that is critical for further protein ubiquitination for building DNA repair foci through protein recruitment and degradation. Bortezomib treatment may prevent DNA resection by inhibiting the proteasomal degradation of proteins involved in chromatin relaxation, thus preventing the recruitment of replication protein A (RPA) onto single-stranded DNA (ssDNA) [44][45][46]. In vitro studies showed that bortezomib can possess synergistic anti-myeloma effects with melphalan by sensitizing MM cells to chemotherapeutic agents [47][48]. In clinical studies, bortezomib in combination with melphalan is efficacious for MM, both in the untreated and relapsed settings [49][50]. In the ASCT setting, the Intergroupe Francophone Du Myeloma (IFM) Phase II study showed significantly better outcomes in the ASCT recipients who received bortezomib–HDM conditioning compared with those treated with HDM only [51]. However, a recent analysis of the IFM 2014-02 trial Phase III study, in which 300 randomized patients received upfront ASCT to bortezomib and HDM, showed no difference in CR rate, PFS, and OS [52]. Doxorubicin is one of the most effective chemotherapy drugs for the treatment of many cancers, including lung cancers, leukemia, lymphoma, and MM [53]. This chemotherapeutic exerts its effects on cancer cells by intercalating into DNA, which results in DNA synthesis inhibition. Moreover, doxorubicin is a DNA topoisomerase II inhibitor, leading to DNA strand breaks by forming a cleavable complex with DNA and DNA topoisomerase II [54] and ROS formation in cells. Alkylating agents and DNA topoisomerase II inhibitors pose the risk of secondary cancers, such as therapy-related acute myeloid leukemia. Compared with de novo acute myeloid leukemia, the therapy-related disease is associated with poor prognosis and abnormal karyotypes, including the −5, −7, abnl(17p), complex karyotypes, and monosomal karyotypes [55].
, were associated with the outcome and overall survival [44]. These genes are significantly enriched in genes involved in homologous recombination repair (HRR) and nucleotide excision repair (NER), necessary for ICL repair. Bortezomib is the first PI approved by the FDA for the treatment of newly diagnosed and relapsed/refractory MM. This drug can bind to and form a complex with the active site of the threonine hydroxyl group in the β5 subunit of the proteasome and block its chymotrypsin-like activity [45]. Recent studies on proteasome inhibition in MM cells have revealed that the accumulation of unfolded proteins in the endoplasmic reticulum, the so-called endoplasmic reticulum stress, triggers that of several pro-apoptotic factors and cell stressors, such as reactive oxygen species (ROS). PI affects DNA repair via depletion of the free ubiquitin pool that is critical for further protein ubiquitination for building DNA repair foci through protein recruitment and degradation. Bortezomib treatment may prevent DNA resection by inhibiting the proteasomal degradation of proteins involved in chromatin relaxation, thus preventing the recruitment of replication protein A (RPA) onto single-stranded DNA (ssDNA) [46,47,48]. In vitro studies showed that bortezomib can possess synergistic anti-myeloma effects with melphalan by sensitizing MM cells to chemotherapeutic agents [49,50]. In clinical studies, bortezomib in combination with melphalan is efficacious for MM, both in the untreated and relapsed settings [51,52]. In the ASCT setting, the Intergroupe Francophone Du Myeloma (IFM) Phase II study showed significantly better outcomes in the ASCT recipients who received bortezomib–HDM conditioning compared with those treated with HDM only [53]. However, a recent analysis of the IFM 2014-02 trial Phase III study, in which 300 randomized patients received upfront ASCT to bortezomib and HDM, showed no difference in CR rate, PFS, and OS [54]. Doxorubicin is one of the most effective chemotherapy drugs for the treatment of many cancers, including lung cancers, leukemia, lymphoma, and MM [55]. This chemotherapeutic exerts its effects on cancer cells by intercalating into DNA, which results in DNA synthesis inhibition. Moreover, doxorubicin is a DNA topoisomerase II inhibitor, leading to DNA strand breaks by forming a cleavable complex with DNA and DNA topoisomerase II [56] and ROS formation in cells. Alkylating agents and DNA topoisomerase II inhibitors pose the risk of secondary cancers, such as therapy-related acute myeloid leukemia. Compared with de novo acute myeloid leukemia, the therapy-related disease is associated with poor prognosis and abnormal karyotypes, including the −5, −7, abnl(17p), complex karyotypes, and monosomal karyotypes [57].
DNA damage requires effective DNA repair capacity, which may be limited in tumor cells. Compared to normal cells, cancer cells present a higher accumulation of DNA damage and replication stress (RS) due to faulty cell cycle checkpoint activation [23][56]. Sources of RS include fragile sites, replication-transcription complex collision, secondary DNA structures, depletion of replication factors and nucleotides, and oncogenic stress [57]. Furthermore, DNA repair activity is required to counteract oxidative DNA damage in tumor cells induced by the tumor microenvironment. Cancer cells utilize mutagenic repair pathways for their advantage and to escape death [23]. Such repair pathways in cancer cells may represent treatment targets for their sensitization to drugs. Indeed, DNA repair inhibition has been used as a strategy for cancer treatment. Therapeutic strategies that target DNA damage include the use of PARP inhibitors for
DNA damage requires effective DNA repair capacity, which may be limited in tumor cells. Compared to normal cells, cancer cells present a higher accumulation of DNA damage and replication stress (RS) due to faulty cell cycle checkpoint activation [23,25]. Sources of RS include fragile sites, replication-transcription complex collision, secondary DNA structures, depletion of replication factors and nucleotides, and oncogenic stress [58]. Furthermore, DNA repair activity is required to counteract oxidative DNA damage in tumor cells induced by the tumor microenvironment. Cancer cells utilize mutagenic repair pathways for their advantage and to escape death [23]. Such repair pathways in cancer cells may represent treatment targets for their sensitization to drugs. Indeed, DNA repair inhibition has been used as a strategy for cancer treatment. Therapeutic strategies that target DNA damage include the use of PARP inhibitors for
BRCA1
/
BRCA2-mutated cancers [58]. Thus, DNA repair pathways, including HRR, may be the target of drugs used to treat MM and play a role in resistance [59][60]. Next, we provide an overview of the DDR and major DNA repair pathways.
-mutated cancers [59]. Thus, DNA repair pathways, including HRR, may be the target of drugs used to treat MM and play a role in resistance [60,61]. Next, we provide an overview of the DDR and major DNA repair pathways.
3. DNA Repair Pathways
Mammalian cells have six major DNA repair pathways involved in the DDR: the base excision repair (BER), NER, and mismatch repair (MMR) pathways repair nucleotide lesions on ssDNA; the HR and NHEJ pathways are involved in DSB repair; and the Fanconi anemia (FA) pathway repairs ICL lesions in co-operation with the NER and HR pathways [23][40][56].
Mammalian cells have six major DNA repair pathways involved in the DDR: the base excision repair (BER), NER, and mismatch repair (MMR) pathways repair nucleotide lesions on ssDNA; the HR and NHEJ pathways are involved in DSB repair; and the Fanconi anemia (FA) pathway repairs ICL lesions in co-operation with the NER and HR pathways [23,24,25].