Polyphenols represent a group of secondary metabolites of plants which have been analyzed as potent regulators of multiple biological processes, including cell proliferation, apoptosis and autophagy, among others. These natural compounds exhibit beneficial effects and protection against inflammation, oxidative stress and related injuries including metabolic diseases, such as cardiovascular damage, obesity, diabetes and neurodegeneration.
- polyphenols
- autophagy
- mechanisms
- oxidative stress
1. Molecular Mechanisms of Autophagic Process and Its Regulation by Dietary Polyphenols
Autophagy is a physiological recycling process highly conserved in eukaryotic cells, playing an important role in maintaining cytoplasmic quality control through the elimination of cytosolic protein aggregates, damaged organelles and invasive microbes and by reducing oxidative and ER stress. This activity aims to maintain cellular homeostasis and cell survival, which contributes to enhance health and longevity [1][2][3][4][35–38]. Moreover, this catabolic process is upregulated by a wide range of extracellular and intracellular stressors such as nutrient starvation including growth factor and insulin deprivation, hypoxia, infections, ER, and oxidative stress; the resulting macromolecular constituents are then recycled. In this context, activation of autophagy allows metabolic adaptation during starvation by providing an alternative source of energy and nutrients [5][39] and participating in the cellular defense against infections [6][7][40,41]. The autophagic process involves the activities of proteins encoded by autophagy-related (ATG) genes and it could be considered as a nonselective degradative pathway, removing non-specific cargoes and ubiquitinated proteins (also referred as “bulk autophagy”), or highly specific, eliminating distinct cargoes including lipids (termed lipophagy), peroxisomes (pexophagy), mitochondria (mitophagy), nucleus (nucleophagy), ribosomes (ribophagy), ER (reticulophagy), and microbes (xenophagy), among others. In mammalian cells, three primary types of autophagy, depending on how the cargo is delivered to the lysosome—macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA)—have been described [8][42]. The present review focuses on the macroautophagy process (hereafter referred to as autophagy) which takes place through the following four steps: (1) initiation, (2) nucleation, (3) elongation, and (4) fusion and degradation [9][10][11][43–45], as described in detail in Figure 12.
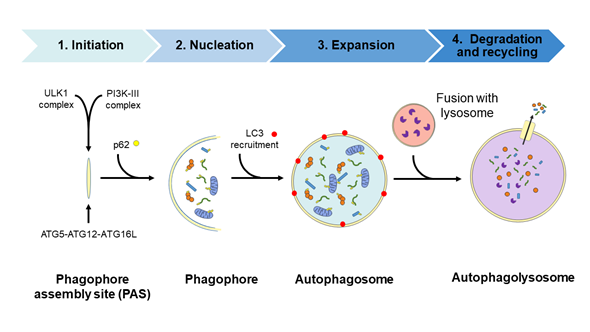
Figure 12. Diagram of the autophagic process. Steps: (1) Initiation, with the formation of the phagophore assembly site (PAS) which assemblies the Atg1/ULK1 complex and the PI3K-III complex; (2) nucleation step occurs upon the recruitment of the ubiquitin-like conjugation system (the ATG5/ATG12/ATG16L complex) to the PAS. Then, the curvature of this double-membrane (phagophore) is induced to facilitate the sequestration of the cargo. The recruitment of the ubiquitinated proteins or damaged organelles to the phagophore is carried out by the p62 protein, which links the autophagy pathway and the ubiquitin-proteasome system (UPS) by binding the ubiquitinated proteins to LC3 protein for autophagic degradation; (3) the elongation of the phagophore to form a spherical vesicle termed autophagosome; (4) finally, autophagosomes fuse with lysosomes to form autophagolysosomes/autolysosomes and the autophagic cargo are degraded by the action of resident lysosomal/vacuolar hydrolases and then are exported to the cytosol for reuse by the cell. ATG, autophagy-related; LC3, microtubule-associated protein 1A/1B-light chain 3; p62, sequestosome 1 (SQSTM1); PI3K-III, class III phosphatidylinositol 3-kinase; ULK1, unc-51-like kinase.
The autophagic process is tightly regulated depending on nutrient status by different signaling pathways that include the mammalian target of rapamycin complex 1 (mTORC1), AMP-activated kinase (AMPK), and Sirtuin-1 (SIRT1) pathways. Autophagy is negatively regulated by the mTORC1 pathway, which also regulates several important and essential processes including cell growth and protein synthesis [12][46]. Under nutrient-rich conditions, mTORC1 is activated and phosphorylates ULK1, thereby preventing its activation by AMP activated protein kinase (AMPK), a key activator of autophagy [13][47]. Moreover, mTORC1 phosphorylates and inhibits the nuclear translocation of the transcription factor TFEB, which drives the expression of genes for lysosomal biogenesis and the autophagy machinery [12][46].
Conversely, mTORC1 is inhibited following nutrient starvation or energy deprivation and, thus, AMPK as a key energy sensor is activated and autophagy is induced [13][47]. When AMPK becomes activated, it phosphorylates and activates the tuberous sclerosis complex (TSC) which is composed of the TSC1 (hamartin), TSC2 (tuberin), and TBC1D7 proteins. The TSC complex is an essential inhibitor of mTORC1 activity through the activation of its GAP (GTPase-activating protein) activity towards the small G-protein Rheb (Ras homologue enriched in brain) [14][48]. Inactivation of mTORC1 activity by the TSC complex occurs via lysosomal recruitment of cytosolic TSC complex, where mTORC1 is located [15][49].
In addition, AMPK activation stimulates the activity of the nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase SIRT1 by elevating the intracellular levels of its co-substrate, NAD+, and inducing lifespan extension [16][50]. Regarding post-translational modifications, the acetylation status of histones as well as cytoplasmic proteins is related with the regulation of autophagic flux [17][18][51,52]. In this sense, growing evidence indicates that both the activity of sirtuins, the deacetylation protein status, and the activation of autophagic flux decline with age [19][53].
Upon starvation, hypoxia, mitochondrial dysfunction, or several other circumstances in which a high intracellular levels of ROS/RSN are achieved, the c-Jun N-terminal kinase (JNK), or stress-activated protein kinase, becomes activated. JNK activation mediates the phosphorylation of B-cell lymphoma 2 (BCL2) protein to induce apoptosis. On the contrary, when JNK is not activated, dephosphorylated BCL2 interacts with beclin-1, which is a BCL2-interacting protein mediating the inhibition of beclin-1—dependent autophagy. This is only one of the main mechanisms of the complex crosstalk between autophagy and apoptosis [20][54].
Mitochondrial quality control mechanisms involved the activity of the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) and an ubiquitin E3 ligase known as Parkin. PINK1 protein acts as a molecular sensor of damaged mitochondria and promotes the recruitment of Parkin on the outer mitochondrial membrane (OMM) resulting in the ubiquitination of numerous OMM proteins, which in turn recruits other proteins to mitochondria to initiate mitophagy. It is well known that alterations of this mitophagy and/or autophagy pathways lead to the accumulation of altered proteins and damaged organelles contributing to oxidative stress.
In addition, another adaptive cellular response and a redox signaling axis that confers cellular protection against oxidative stress is the nuclear factor erythroid 2-related factor 2 (Nrf2)-Kelch-like ECH-associated protein 1 (Keap1)-antioxidant response elements (ARE) pathway. Activated Nrf2 is translocated to the nucleus where it activates the transcription of antioxidant genes, including heme oxygenase-1 (HO-1) [21][55]. Of note, and regarding the crosstalk between the antioxidant effects mediated by Nrf2-Keap1-ARE activation and autophagic process, it has been described that p62/SQSTM1 protein, which is degraded by autophagic process, also represents one of the Nrf2 target. Moreover, p62/SQSTM1 protein competitively binds to the redox sensor Keap1, disrupting its interaction with Nrf2, resulting in Nrf2 activation [22][56]. As a consequence, this p62/SQSTM1 positive-feedback loop may have beneficial effects against oxidative stress-dependent cell death through the activation of mitophagy by increasing the degradation of defective and altered mitochondria [4][38]. Other mechanisms are involved in autophagy regulation by oxidative stress such as post-translational modifications of key autophagy regulators. In this sense, recent studies demonstrate that some plant polyphenols including resveratrol, curcumin, and quercetin are able to activate the Nrf2-Keap1-ARE, counteracting oxidative damage and representing a novel therapeutic approach based on the antioxidant properties of these natural compounds[23][24] [29,57,58].
Moreover, there has been a growing interest in the beneficial effects of the dietary consumption of polyphenols beyond its antioxidant activity, because most of them are able to activate autophagy and, thus, play a beneficial role in the redox balance by alleviating oxidative stress. Therefore, dietary polyphenols have emerged as a promising therapeutic strategy to prevent the development of several diseases including metabolic and neurodegenerative ones [25][57,59].
Resveratrol (2,3,4′-trihydroxystilbene), which is a caloric restriction mimetic (CRM), stimulates in vivo SIRT1, inducing the kinase activities of liver kinase B1 (LKB1) and AMPK, and inhibiting mTORC1 signaling, leading to autophagy activation [26][60]. In addition, resveratrol reduces ROS production in response to D-galactose of aging cardiomyocytes, enhancing cardiac function and diminishing ischemia/reperfusion-induced cell apoptosis through the regulation of mitophagy [27][61]. Moreover, resveratrol could enhance the association between mTORC1 and its inhibitory protein DEPTOR (DEP-domain containing mTOR-interacting protein), thus triggering the inactivation of mTORC1 [28][62]. It also increases in vivo mitochondrial biogenesis and function [26][60].
Flavonoids such as fisetin and quercetin lead to autophagy induction by TFEB nuclear translocation via mTORC1 inhibition [29][30][63,64]. The polyphenol curcumin is able to translocate TFEB to the nucleus, leading to autophagy activation by mTORC1-independent inhibition [31][65]. In addition, curcumin activates AMPK, which in turn phosphorylates BCL2, and subsequently disrupts the interaction between BCL2 and BECN1, resulting in autophagy activation [32][66] or upregulation of the expression of BECN1 protein which reduces p62 levels [33][67]. More recently, it has been demonstrated that the dietary tannin punicalagin activates autophagy through the inhibition of Akt signaling pathway, resulting in the activation of the transcription factor forkhead box O3a (FOXO3a) [34][68]. FOXO transcription factors have been identified to promote the expression of ATG genes, including LC3b (Map1lc3b), Gabarapl1, Pi3kIII, Ulk2, Atg12l, Beclin1, Atg4b, Atg14, and Bnip3, then leading to an increase in both autophagic and mitophagic flux [35][69].
Oleuropein and its related compounds, which are secoiridoids present in olive oil, have also emerged as autophagic inductors because they could activate the Ca2+/CaMKKβ/AMPK signaling pathway and hence preserve TFEB in its activated state [36][70] or even induce the expression of proteins involved in autophagic process including BECN1 and LC3 [37][38][71,72] mainly caused by AMPK activation [39][73]. Besides all these specific mechanisms, both mono- and polyphenols could also activate autophagic flux by stimulating the deacetylase activity of SIRT1 and, consequently, downregulating the general acetylation status of cytosolic proteins [40][74].
2. Dietary Polyphenols and Type 2 Diabetes, Obesity and Metabolic Syndrome
The mechanism of action of different polyphenols in the control of several metabolic diseases, including obesity and type 2 diabetes mellitus (T2DM) in relation with autophagy modulation, has been extensively studied. It is well known that polyphenols are a group of molecules with positive effects in the modulation of aging, a period in which most of the reviewed diseases show a higher prevalence. T2DM is a very complex disease and is considered as epidemic worldwide. It is the resultant from multiple factors in a progressive manner, being insulin resistance and pancreatic β cell dysfunction the most relevant. T2DM is associated with increased levels of glucose and lipids that could contribute to β-cell death and is characterized by two different phases. During the first one, which main event is insulin resistance with normal levels of glycemia, pancreatic β-cells increase their mass by hyperplasia or hypertrophy, with a concomitant insulin and amylin secretion [41][75]. The duration of this phase depends on the patient’s idiosyncrasy and can be extended for years. At the final stage, pancreatic β-cells fail and then hypoinsulinemia appears. In addition, amylin deposition occurs in nearly 90% of T2DM patients. During the progression to T2DM, a chronic activation of mTORC1 signaling pathway has been detected. Although this activation is necessary during the first phase of the disease, for insulin resistance compensation by pancreatic β-cells, its chronic effect is deleterious for these cells [42][43][76,77]. Importantly, this maintained activation of mTORC1 generates a blockade in autophagic flux, which is essential for a correct elimination of damaged organelles or protein aggregates. These effects generate a continuous stimulation of the unfolded protein response (UPR), which in turn upregulates ER protein chaperones in charge of promoting protein folding. However, when the ER protein folding capacity is overwhelmed, cells undergo a condition of chronic ER stress, triggering the activation of apoptosis [44][45][78,79]. Nowadays, T2DM is considered a disease affecting the folding ability of pancreatic β-cells. In fact, the expression of different endogenous ER chaperones such as the 78-kDa glucose-regulated protein (GRP78) or BiP protein and protein disulfide isomerase, or chemical chaperones, such as taurine-conjugated derivative from ursodeoxycholic acid (TUDCA) or 4-phenyl butyric acid (4-PBA), diminished β cell failure and facilitated the correct folding, avoiding protein aggregation and apoptosis [46][80]. In this regard, azoramide, a small molecule which modulates UPR activity, exerts a strong antidiabetic activity [47][48][81,82]. In addition to all the changes mentioned before through the progression to T2DM, mitochondrial dysfunction is also shown. In general terms, mitochondrial dysfunction occurs as the natural history of aging [49][83]. However, during T2DM progression a concomitant defect in the mitochondrial clearance or mitophagy occurs which promotes an accumulation of aberrant and dysfunctional mitochondria which cannot be eliminated by mitophagy [50][84]. In this regard, a chronic activation of mTORC1 in pancreatic β-cells is able to inhibit both general autophagy as well as mitophagy, with a higher production of oxidized mitochondrial proteins and an accumulation of mitochondria with an altered membrane potential, which are not degraded by mitophagy [51][85]. One of the mechanisms that could explain this failure in mitophagy is the reduction in the levels of PINK1, which is an essential component in the recognition of mitochondria with an altered membrane potential as observed in fibroblasts with mTORC1 hyperactivation [52][86]. In addition to that, the overexpression of human amylin in pancreatic β cells impairs both bulk autophagy as well as mitophagy [53][87]. More importantly, these defects have been observed in prediabetic stages as well as in T2DM patients [54][88].
One of the most characterized and studied polyphenols with respect to its effects in diabetes is the stilbene compound resveratrol. Apart from its protective effects in diabetes, resveratrol can be a positive treatment in diabetes-related pathologies such as diabetic cardiomyopathy [55][89]. In fact, positive effects of resveratrol in the reduction of oxidative stress [56][90] as well as an increase in the sensitivity to insulin [57][91] with a concomitant decrease in lipid levels have been assessed [58][92]. In addition to these actions, resveratrol has been proposed to modulate the expression of both pro-apoptotic and anti-apoptotic factors [59][31]. Furthermore, stilbenes can stimulate an important antioxidant defense such as the transcription factor called Nrf2. Nrf2 is involved in the control of the transcription of different antioxidants in response to inflammation and oxidative stress [60][93]. Stilbenes are also involved in the activation of autophagy, through the modulation of p62 protein levels and SIRT1 activity [61][94]. An essential role of SIRT1 has been proposed for the induction of autophagy in response to hypoxia in a T2DM rat model [62][95]. Resveratrol stimulates autophagic flux, improving diabetic cardiomyopathy in a diabetic mouse model mediated by a decrease in p62 protein levels, facilitating SIRT1 activity. Moreover, SIRT1/FOXO/Rab7 has been proposed as a potential therapeutic pathway which mediates the effects of resveratrol on autophagic flux and ameliorates dysfunctional autophagy in diabetic cardiomyopathy [62][96]. Resveratrol can also modulate autophagy by alternative indirect mechanisms which involve transcriptional regulation of microRNA´s (miRNA´s for short). miRNA-18a-5p is one kind of miRNA which expression is enhanced in a diabetic mouse model after treatment with resveratrol [63][97].
Other polyphenols with a wide protective effect in diabetes are curcumin and its analogues. These groups of molecules are able to down-regulate the chronic stimulation of ER stress, reducing apoptosis [64][98] and increasing insulin sensitivity [65][99]. There are multiple evidences indicating a pro-autophagic role of curcumin in order to moderate the negative effects of T2DM [32][66]. For instance, curcumin can modulate several proteins involved in autophagy including LC3, p62, and beclin-1, among others. These effects have been described in a diabetic mice model treated with curcumin, preventing podocyte apoptosis during diabetic nephropathy [66][67].
(-)-Epigallocatechin-3-gallate (EGCG) is a polyphenol isolated from green tea with a significant effect in autophagy modulation. EGCG is able to regulate autophagy at different levels, alleviating part of the deleterious effects observed in diabetic patients. For instance, it can avoid lipid accumulation in vascular endothelial cells, increasing the degradation of lipids by lipophagy, and facilitating the recognition between LC3B (located in the autophagosomes) and the lipid droplets for their catabolism [67][100]. This polyphenol has a protective effect for both mitochondrial dysfunction and aberrant autophagy described in the heart’s tissue of diabetic rats [68][69][101,102].
Punicalagin, one of the main polyphenolic components of pomegranate, is able to protect liver dysregulation in response to T2DM. In fact, this molecule has been proposed very recently as an autophagy inducer through the modulation of different protein markers such as LC3B and p62 which allow the reactivating of autophagy [34][68]. Furthermore, oleuropein has been related to an induction of autophagy as part of its mechanism of action [39][73]. Oleuropein, together with hydroxytyrosol, is involved in the induction of autophagy through the regulation of AMPK/mTORC1 signaling pathway [70][70,103].
Obesity, one of the main factors associated to the development of T2DM, has been implicated in autophagy dysregulation. In fact, a dysregulation in autophagic activity plays an important role in the appearance of insulin resistance [71][104]. Furthermore, adipocyte hypertrophy occurring in obesity contributes to insulin resistance as well, by the production of different inflammatory cytokines which lead to an increased in endoplasmic reticulum stress (ER-stress) [72][105]. One of the main polyphenols with protective properties in obesity and, specifically in liver steatosis, is resveratrol. In rats, resveratrol effect is similar to an energy restriction (a reduction of 15% of calories in energy intake), with a decrease in p62 protein levels and an increase in LC3B, beclin-1, and atg5 protein levels, which collectively suggests an autophagic activation [73][106]. Apart from resveratrol, other polyphenolic compounds with protective effects through autophagy activation in obesity have been assessed. A treatment with epigallocatechin-3-gallate (EGCG) for two weeks in a mice model of high-fat diet (HFD)-induced obesity stimulated both autophagic flux in white adipose tissue in an AMPK-dependent manner and beclin-1 activation [74][107]. Very interestingly, the effect of a 30 days’ supplementation with resveratrol on gene expression and adipose tissue morphology in a cohort of obese men has been reported. The authors indicated that, at the end of the study, there was a significant reduction in adipocyte size, with a downregulation of both Wnt and Notch signaling pathways. In addition, there was an upregulation in different proteins involved in the cell cycle control which suggested an increase in adipogenesis. Finally, an increase in lipophagy accompanied by a reduction in inflammation were also observed [75][108].
The metabolic syndrome represents several injuries including obesity, T2DM, and nonalcoholic fatty liver (NAFL); a mitochondrial dysfunction appears, thus contributing to insulin resistance. Then, mitophagy could potentially preserve mitochondrial function by the elimination of these damaged organelles [76][77][109,110]. Another important consequence of obesity is the appearance of NAFLD. It is well known that the elimination of lipid accumulation that occurs in the liver by the activation of lipophagy contributes to a decrease in NAFLD pathology [78][111]. In this regard, there are multiple examples assessing the protective effect of different polyphenols: pro-autophagic activity of resveratrol has been demonstrated both in vitro model of palmitic acid-induced hepatic steatosis [79][112] and in in vivo models of NAFLD, where it also reduces liver inflammation [113,114]. In fact, this in vivo beneficial effect of resveratrol was associated with autophagy activation, because the elimination of ULK1, which is essential in the control of autophagosome formation, impaired the protective role of resveratrol [80][113]. Using a cafeteria diet-induced obesity in rats, polyphenols from bergamot were able to induce lipophagy and prevent NAFLD onset. In the bergamot-treated animals, a reduction in p62 protein levels and an increase in both LC3B and beclin-1 protein levels were shown, both events being associated with an activation of the autophagic flux. All these effects were linked to a decrease in both liver lipid accumulation and inflammation [81][82][115,116]. Furthermore, an in vitro model of liver cells (HepG2) treated with different levels of fatty acids (a combination of oleic acid and palmitic acid) which are found in patients with liver steatosis, assessed the accumulation of lipids inside the cells. Very interestingly, the protective effect of different polyphenols derived from Toona sinensis A. Juss. was associated with an activation of AMPK/ULK1 signaling pathway with a concomitant increase in LC3B protein levels[83] [117]. Similar protective results related to lipid accumulation in a mice model of NAFLD in response to blueberry polyphenols were reported [84][118]. Very recently, the protective effect exerted by a polyphenol extract from apple in the activation of lipophagy and restoration of lysosomal pH was related to intervention through the SIRT1/AMPK signaling [85][119]. In fact, polyphenols such as quercetin can stimulate lysosomal biogenesis and, hence, the clearance capacity of the cells by facilitating the nuclear translocation of TFEB [30][64]. Although these effects have been observed in retinal epithelium cells (RPE cells), it should be of great interest to explore the improvement of the TFEB-lysosomal axis in other pathologies such as T2DM, obesity, and metabolic syndrome. In summary, autophagy activation represents a key event in the pathophysiology of fatty liver disease and its modulation by the use of different polyphenols could contribute to prevent both the disease and related pathologies such as T2DM, obesity, and other metabolic complications [86][120].
3. Effect of Polyphenols on Cardiovascular Diseases
Cardiovascular diseases are a direct consequence of aging. During the aging process, there is a progressive deterioration of mitochondrial function and a decline in cardiac efficiency. Using a cellular model of senescent-like cardiomyocytes, treatment with resveratrol improved cardiac function by the activation of SIRT1 signaling pathway. In addition, the effect of resveratrol in the correct function of two essential proteins in mitophagy namely PINK1 and PARKIN was also assessed [27][61]. A polyphenol mixture from green tea (catechin > 90% and EGCG > 70%) exerted a protective role in the reduction of atherogenesis in an ApoE-knock-out mice model through the activation of autophagy. In this model, there was an increased production of p62 protein levels linked to an increased autophagosome formation flux, with an increase in LC3B and beclin-1 protein levels in the polyphenol-treated group [87][121]. Oleuropein aglycone has been studied in a cellular model with an increased oxidative stress in cardiomyocytes. In these cells, there was an induction of autophagy, detected by an increase in LC3B and beclin-1 protein levels. However, very importantly, there was an improvement of TFEB translocation to the nucleus which suggested an improved autophagy induction [38][72]. In this regard, different polyphenols have been involved in the protection against cardiovascular disease and vascular inflammation, including EGCG, quercetin, resveratrol, apigenin, and curcumin, through autophagy modulation among other mechanisms [88][89][122,123]. Recent reports indicate that resveratrol, by autophagy activation could be useful for the treatment of the ischemic process found in diabetic myocardium. The beneficial effect of resveratrol was associated with an increase in beclin-1 and LC3B protein levels and a reduction in inflammation, by diminishing two inflammatory cytokines such as TNF-alpha and IL-6 [90][124]. In general terms, mTOR signaling pathway has been proposed as a target for treating atherosclerosis, cardiac hypertrophy, and heart failure. Then, every natural compound with proven activity towards the inhibition of mTORC1, could represent an interesting approach to reduce both the above cited diseases and the number of deaths caused by cardiovascular disease worldwide [91][125].