The human P2X7 gene is highly polymorphic, and several splice variants of the receptor have been identified in time. P2X7 single-nucleotide polymorphisms (SNPs) have been broadly analyzed by studies relating them to pathologies as different as infectious, inflammatory, nervous, and bone diseases, among which cancer is included. Moreover, in the last years, an increasing number of reports concentrated on P2X7 splice variants’ different roles and their implications in pathological conditions, including oncogenesis. Our article on P2X7 variants in oncogenesis recently published in Cells gives an overview of established and recent literature demonstrating a role for human P2X7 gene products in oncological conditions. Here you find the main structural information on P2X7 variants.
- P2X7
- P2X7 splice variants
- P2X7 SNPs
- P2X7B
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
P2 purinergic receptors are plasma membrane proteins activated by nucleotides and subdivided into metabotropic P2Y and ionotropic P2X receptors. P2X receptors are ATP gated ion channels permeable to Na+, K+, and Ca2+ that assemble as homo or heterotrimers of seven different subunits (P2X1-7) [1]. The P2X7 subunit, with its 595 amino acids (aa), is the most extended protein in the P2X family, and it assembles to form a homo-trimeric receptor. Different domains form the P2X7 structure, including a short intracellular N-terminal tail (26 aa), a large extracellular loop (282 aa) containing the ATP binding site, two transmembrane helices (24 aa each), and a long cytoplasmic carboxy-terminal tail (239 aa) [2][3][2,3]. Among these domains, the C terminal tail is unique at P2X7, as it is not present in any other member of the P2X family, and it confers to the receptor the ability to interact with several intra-cytoplasmic and transmembrane proteins [4][5][6][4–6]. Moreover, the P2X7 C-tail is responsible for a peculiar activity triggered by the receptor: the opening of a big non-selective pore, which is known as the “macropore” [7][8][2,7,8]. The macropore is activated upon a long exposure of P2X7 to high concentrations of its ligand ATP, and it is permeable to aqueous solutes such as ethidium bromide, propidium iodide, YO-pro, and Lucifer Yellow [7]. The macropore formation is reversible, as the removal of ATP, in a few minutes from its addition, induces the reconstitution of cell membrane integrity [9]. Whether the macropore-related activities are dependent only on P2X7 permeabilization or also on accessory pathways or intracellular signaling is still an object of debate [10][11][10,11]. However, it is well accepted that the macropore’s opening is associated with cytotoxicity; instead, the tonic activation of P2X7 with limited ATP stimulation supports a trophic effect [12]. While the general tertiary structure of the P2X7 is comparable to that of other P2X receptors [13][14][15][13–15], that of the C terminal tail was missing until a recent study by McCarthy et al. [16]. The authors reported the presence of a C-cys anchor domain that, if palmitoylated, causes the typical non-desensitizing current associated with P2X7. Based on this molecular structure, a following study proposed that the P2X7 C terminal domain was acquired by genomic rearrangement from a P2X4-like gene in ancient jawed vertebrates generating the actual mammalian P2X7 [17]. Therefore, it is not completely surprising that some P2X7 splice variants, which function as a channel but not as macropore have been conserved through evolution [18]. Many cell types and tissues express the P2X7 receptor, but its role was mainly studied in the immune system, where its activity is associated with inflammatory conditions [19][20][19,20]. Indeed, P2X7-dependent activation of the NLRP3 inflammasome and caspase 1 and the consequent maturation of pro-IL1β is an established notion [21]. The P2X7 receptor is also central to the cytokine’s vesicular release from dendritic cells, macrophages, and microglia [22][23][24][22–24]. The implications of P2X7 activity in inflammatory and immune disorders, neuronal, cardiovascular, and metabolic diseases, and cancer have been recently appraised in several reviews [25][26][27][28][29][30][31][32][14,25–32].
2. Human P2X7 Splice Variants and Single-Nucleotide Polymorphisms
The human P2X7 receptor gene is located on chromosome 12 and consists of 13 exons [33]; it is highly polymorphic and gives rise to several splice variants [34]. The splice variants, which were initially identified around 2005, are listed as (P2X7A-J) [35][18,35], where P2X7A is the mRNA coding for the complete protein and generally referred to as a full-length P2X7 receptor [36]. Among the other alternative mRNAs, P2X7B, P2X7E, P2X7G, and P2X7J give rise to truncated proteins that lack the extended C-terminal tail [35][18,35]. The splicing isoforms are generated either via the inclusion of a new exon or the exclusion of one or more genetic regions. Indeed, P2X7G and P2X7H have an inserted additional exon, while P2X7C, P2X7D, P2X7E, and P2X7F lack respectively exon 4, exon 5, exons 7 and 8, exons 4 and 8 [36][18,36]. Instead, P2X7I derives from a point mutation in the first intron of P2X7, leading to an extended truncation, and P2X7J is truncated after exon 7, making it non-functional (Figures 1 and 2). Nevertheless, it acts as a dominant-negative on P2X7A [35]. On the contrary, P2X7B exerts a dominant positive activity at the main isoform [8,37]. The P2X7B variant was probably the most studied, as it is the only one to be functional as a small ion channel [37][38][39][40][41][42][8,18,37–42]. The P2X7B isoform retains an intron between exons 10 and 11, causing the addition of 18 extra amino acids after residue 346, which is followed by a stop codon [18] (Figure 1). Nevertheless, P2X7B EC50 is relatively high, and depending upon the cell type and experimental conditions tested, it is not always expressed and functional at the plasma membrane [41][8,37,41]. Another recently identified alternative mRNA for the human P2X7 is P2X7-V3 (Figure 1). This splice variant does not give rise to a functional protein but to a long non-coding RNA endowed with a protumoral activity [43]. Another conformational form of the receptor losing P2X7-dependent macropore formation is nfP2X7. In this case, currently, there is no reported sequence allowing us to define whether it is a splice variant or a polymorphic form of the receptor. However, we know that nfP2X7 is recognized by polyclonal antibodies raised against a peptide of human P2X7, including amino acids 200–216. This sequence is accessible as an antigen only in nfP2X7 and not on the wild-type receptor [44]. siRNA experiments have demonstrated that nfP2X7 is a product of the endogenous human P2X7 gene, thus strongly suggesting that this conformational variant could be originating from either alternative splicing or point mutations [45]. nfP2X7 is generally present as an intra-cytosolic protein, which can be translocated at the plasma membrane when the cells are exposed to high ATP concentrations, such as those present in the tumor microenvironment [45]. Similar to P2X7B, nfP2X7 is active as a small ion channel but not as an ethidium permeable macropore; however, unlike P2X7B, it does not exert a dominant positive effect on P2X7A [45].
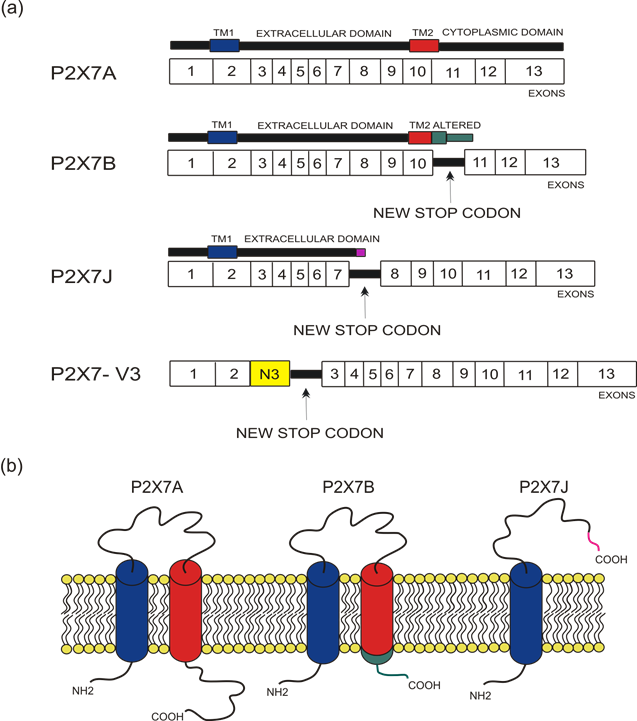
Figure 1. Depiction of human P2X7 receptor splicing variants described in this overview as involved in carcinogenesis and for which a published sequence is available. Different colors identify each region: transmembrane domain 1 (TM1) is in blue, transmembrane domain 2 (TM2) is in red, altered carboxy-terminal tails of P2X7B and P2X7J are respectively in green and in purple. (a) Schematic representation of the human P2X7 gene and its splice variants. The boxes represent the 13 exons, while the black lines between exons represent new introns. The full-length P2X7A variant derives from the original sequence formed by 13 exons. The P2X7B isoform is a truncated variant as it retains an intron between exons 10 and 11, which includes a new stop codon altering the carboxy-terminal tail. P2X7J is a truncated isoform that lacks exon 8 and carries a modified carboxy-terminal tail. P2X7-V3 gains an extra exon called N3 (yellow) and a stop codon; it does not give rise to a protein but instead acts as a short non-coding RNA. (b) Schematic representation of the full-length P2X7A and the truncated isoforms P2X7B and P2X7J expressed on the cell membrane.
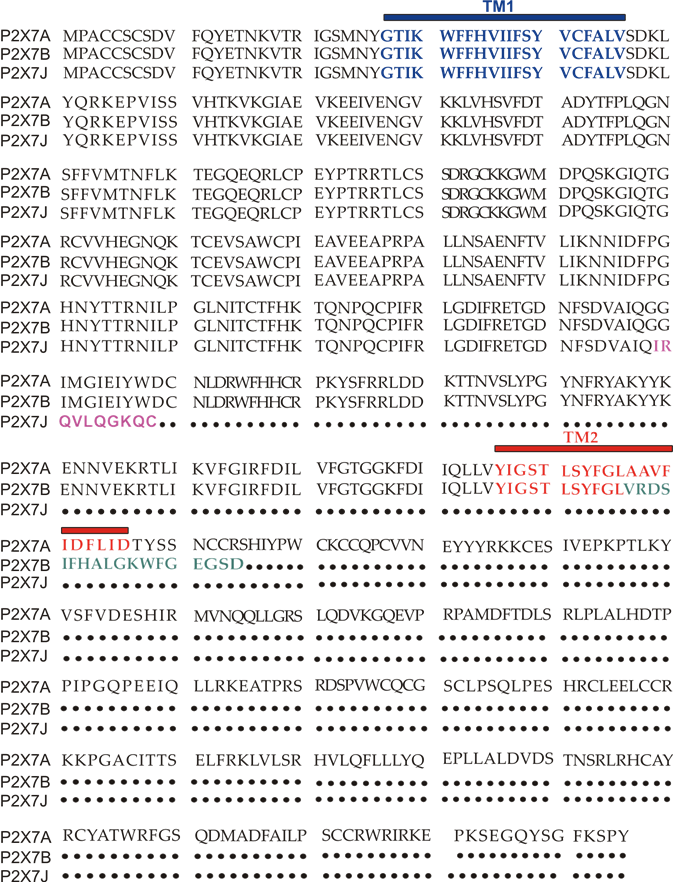
Figure 2. P2X7A(full length) and P2X7B and J amino acids sequence alignment. TM1 is in blue, while TM2 is in red. The ten aminoacids unique to the P2X7J isoform are in purple, while the 18 extra amino acids characterizing P2X7B and located after TM2 are in green.
More than 13,000 single-nucleotide polymorphisms (SNPs) of the P2X7 receptor were identified in the human gene. Among those SNPs, 10 were demonstrated to give rise to loss-of-function variants, and three were demonstrated to give rise to gain-of-function variants [46][34,36,46]. This significant variability was possibly due to the receptor’s central role in response to infection and other inflammatory conditions [19,20]. Several studies associated P2X7 SNPs with the predisposition to arise different diseases, influence cell functionality, and induce cancer development [34]. Here, we describe the characteristics of the SNPs that, to our knowledge, were so far associated with oncological conditions (Figure 3). The first human P2X7 SNP to be identified was the loss of function 1513A > C (rs3751143) variant characterized by the substitution of glutamic acid with alanine in position 496 (E496A) in the long C terminal tail of the receptor [47] (Figure 3). Experiments conducted on leukocytes homozygotes for this SNP revealed a nearly complete loss of activity both as a channel and macropore [47]. The effects of this SNP expression have been extensively studied in different cell types and were associated with several cell dysfunctions that involve the release of pro-inflammatory cytokines, cell death, and membrane molecules exposure [48]. These impairments were associated with susceptibility to tuberculosis infection in different populations [48], a high risk of sporadic Parkinson’s development in a Chinese population [49], and with the increase of bone fracture events in postmenopausal women [50]. As P2X7 1513A > C variant, also, the 946G>A (rs28360457) polymorphism causing the substitution of the arginine at amino acid position 307 with glutamine (R307Q) leads to a loss of function by changing the site of ATP interaction in the extracellular domain [51] (Figure 3). Similarly, the 835G > A (rs 7958311) SNP causes the substitution of histidine 270, which is located in the extracellular domain of P2X7, with an arginine (H270R), and the 1096G > C variant (rs2230911) leads to a serine to threonine change in position 357 of the carboxy-terminal tail (T357S) and both mutations cause P2X7 loss of function [52] (Figure 3).
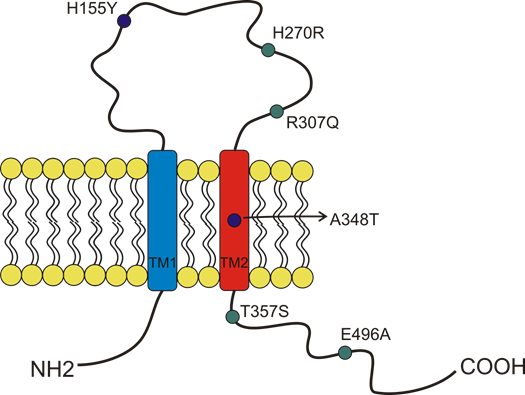
Figure 3. Schematic representation of the position of P2X7 receptor single-nucleotide polymorphisms (SNP) studied in cancer and covered by this overview. The gain of function SNPs are in blue, while the loss of function SNPs are in green.
Among the three gain-of-function polymorphisms known, the most recurrent P2X7 SNP in humans is the 489 C > T (rs208294), which carries a tyrosine instead of a histidine at position 155 in the extracellular domain (H155Y) [53] (Figure 2). This SNP increases the receptor function as both an ion channel and macropore and, interestingly, it corresponds to the wild-type allele in the mouse and rat P2X7, which is long known to have lower EC50 for agonists than the human isoform [54][2,33,54]. Hu and colleagues demonstrated that this polymorphism is associated with the activation of NLRP3 inflammasome and causes a rise of IL-1β and IL-18 secretion [55]. The notion that pro-inflammatory molecules’ secretion is one of the causes that can predispose to mood disorders and inflammatory diseases is an established one. Indeed, human P2X7 489 C > T SNP was associated with pathological conditions such as Alzheimer’s [56], Systemic Lupus erythematosus [55], HHV-6A infection, and related infertility [57], as well as cancer pain [58]. Moreover, it was co-associated with another gain-of-function SNP, the 1068G > A (rs17118119), with increased pain sensation in females affected by diabetic peripheral neuropathic pain [59]. The 1068G > A SNP gives rise to a protein characterized by the presence of threonine instead of alanine at amino acid position 348 (A348T), which is located at the second transmembrane domain. Monocytes carrying the 1068G > A SNP secreted 3-fold more IL-1β than those collected from P2X7 wt subjects when treated with LPS. Threonine 348 also increased P2X7-mediated ethidium uptake and Rb(+) efflux [60]. Based on these data, Stokes and colleagues proposed that this SNP could be a predisposing factor for the development of inflammatory, infectious, and psychiatric disorders [60]. The third gain-of-function variant discovered is the 523C > G, in which the arginine in position 166 of the extracellular domain is substituted by glycine (A166G) [61].