Resistance to anticancer treatments poses continuing challenges to oncology researchers and clinicians. The underlying mechanisms are complex and multifactorial. However, the immunologically “cold” tumor microenvironment (TME) has recently emerged as one of the critical players in cancer progression and therapeutic resistance. Therefore, TME modulation through induction of an immunological switch towards inflammation (“warming up”) is among the leading approaches in modern oncology. Oncolytic viruses (OVs) are seen today not merely as tumor cell-killing (oncolytic) agents, but also as cancer therapeutics with multimodal antitumor action. Due to their intrinsic or engineered capacity for overcoming immune escape mechanisms, warming up the TME and promoting antitumor immune responses, OVs hold the potential for creating a proinflammatory background, which may in turn facilitate the action of other (immunomodulating) drugs. This review deals with the smallest among all OVs, the H-1 parvovirus (H-1PV), and focuses on H-1PV-based combinatorial approaches, whose efficiency has been proven in preclinical and/or clinical settings. Special focus is given to cancer types with most devastating impact on life expectancy that urgently call for novel therapies.
- parvovirus
- oncolytic
- tumor microenvironment
- immunotherapy
- combination therapy
- glioblastoma
- pancreatic cancer
- colorectal cancer
- melanoma
1. Introduction
The rodent H-1 protoparvovirus (H-1PV) (for an overview of H-1PV classification and biology, we redirect the readers to a recent article by Bretscher and Marchini [1]) was first discovered as a contaminating agent in xenotransplanted human tumor cell lines. Originally identified as a pathogen, which lethally affects rat fetuses and newborn rats by causing cerebellar hypoplasia and hepatitis, H-1PV was later found to preferentially replicate in rat and in human transformed or tumor-derived cell cultures, while sparing their non-malignant counterparts [2]. H-1PV intrinsic oncotropism and oncoselectivity are a complex phenomenon based on multiple molecular determinants, which are underrepresented in normal cells, but characteristic of tumor cells. Importantly, humans are not naturally infected with this virus, and no association between H-1PV and human disease has been observed. Two early clinical studies of virus administration to cancer patients—dating back to the 1960s and 1990s of the last century—demonstrated the lack of H-1PV pathogenic effects and the feasibility of the approach, thus laying the groundwork for the development of parvovirus (PV)-based oncolytic virotherapy. Three decades of laboratory efforts yielded extensive preclinical evidence of H-1PV broad tumor-suppressive potential. Furthermore, it became increasingly apparent that in addition to directly inducing cancer cell death (oncolysis), H-1PV was also capable of exerting immunostimulating effects in various preclinical cancer models.
PV-induced immune system stimulation results from multiple infection-associated immunogenic events. Depending on the tumor model, virus dose, route of administration and the immunological status of the host, one or another immunogenic stimulus may prevail. Regardless of the particular mechanism involved, PV-mediated immunomodulation contributes to the “warming up” of the tumor microenvironment (TME) (Figure 1), increases tumor visibility and enhances immune cell reactivity. H-1PV infection-associated immunogenic events and their impact on the immune system are reviewed in detail elsewhere [3].
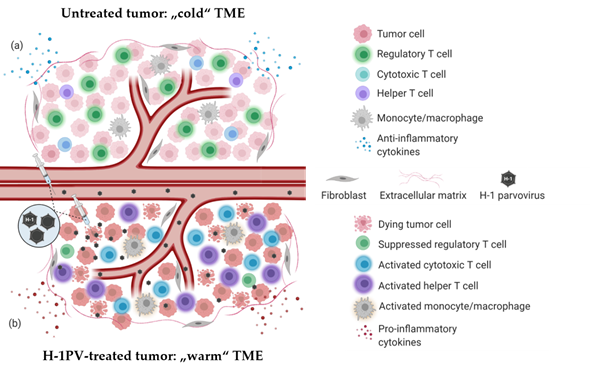
Figure 1. H-1PV-induced modulation of tumor microenvironment immune landscape. (a) Immunosuppressive (“cold”) tumor microenvironment (TME) of a solid tumor. The tumor is often infiltrated with immunosuppressive regulatory T cells (Treg)/myeloid-derived suppressor cells (MDSC). Tumor-infiltrating lymphocytes (TILs) (CD8+ CTLs, CD4+ Th cells) are scarce and/or anergic. Tumor and various TME cells produce anti-inflammatory cytokines to maintain immune suppression and facilitate tumor growth and dissemination. (b) Tumor infection with H-1PV results in immunogenic tumor cell death leading to the release of proinflammatory cytokines, pathogen- and danger-associated molecular patterns (PAMPs and DAMPs), which alarm the immune system. The infection of tumor cells does not necessarily have to be productive for this immuno-stimulating effect to be achieved. Furthermore, abortive infection of immunocytes (CTLs, Th cells, monocytes/macrophages) with H-1PV can also lead to their activation. In contrast to the latter, H-1PV inhibits the immune suppressive functions of Treg cells. An immunological switch takes place and converts the “cold” TME into a “warmed up” (inflamed) one. Virus-mediated immunoconversion of TME favors the mounting of enhanced antitumor immune responses.
The above-outlined H-1PV potential for creating a proinflammatory immune environment and alerting the immune system to the presence of a tumor opens prospects for combining the virus with various immunomodulators or other therapeutic agents endowed with immuno-stimulating properties. This combinatorial approach is in particular promising for the treatment of human tumors that remain presently incurable and pose continuing research and clinical challenges. Pancreatic ductal adenocarcinoma (PDAC), glioblastoma, colorectal cancer (CRC) and melanoma are among those cancers, which are urgently calling for novel therapeutic strategies. H-1PV-based immunotherapeutic combinations are reviewed below, which aim at targeting these devastating malignancies.
Parvovirus-Based Combinatorial Immunotherapy against Pancreatic Cancer
2. Parvovirus-Based Combinatorial Immunotherapy against Pancreatic Cancer
H-1PV is among the oncolytic viruses (OVs), which have promising potential for efficiently targeting pancreatic cancer. PDAC sensitivity to H-1PV-induced oncolysis was demonstrated in various preclinical models [4]. Infection of human PDAC-derived cells leads to their killing, mediated at least in part by cathepsins. Importantly, H-1PV sensitivity is preserved in gemcitabine-resistant cultures, thus opening up prospects to circumvent PDAC resistance to current standard death inducers.
H-1PV + Nucleoside Analogues (Gemcitabine)
As gemcitabine is currently considered the gold chemotherapeutic standard in PDAC clinical management, the therapeutic efficacy of gemcitabine in combination with H-1PV was tested in a rat syngeneic orthotopic PDAC model. H-1PV administration to gemcitabine-pretreated animals led to significant tumor suppression and survival prolongation in comparison with the mock-infected or gemcitabine-only treated groups. These in vivo findings could not be straightforwardly ascribed to synergistic tumor cell death enhancement only. Indeed, in vitro studies showed that the cytotoxic effects of the combination, while allowing effective dose reduction for both agents, did not result in complete PDAC culture elimination. This prompted the investigation of the immunological effects exerted by the H-1PV+gemcitabine combination as an added value to direct tumor cell killing. Markers of immunogenic cell death (ICD) induction were analyzed in various PDAC cell lines, treated with either virus (or gemcitabine) alone or with H-1PV+gemcitabine. It was demonstrated that the release of high-mobility group box 1 protein (HMGB1) is a strikingly robust feature of H-1PV-infected PDAC cells. Furthermore, H-1PV-triggered HMGB1 release did not require lytic infection, in line with the previously reported PBMC activation by non-productively infected PDAC cells. Gemcitabine alone was unable to induce HMGB1 secretion, yet H-1PV-induced HMGB1 release remained unaffected in gemcitabine-treated cells. Gemcitabine, on the other hand, was able to induce—albeit not in all cell lines tested—mature interleukin 1-beta (IL-1ß) accumulation in culture supernatants. Taken together, these data show that H-1PV and gemcitabine complement each other in the induction of immunogenic signals. The compatibility of H-1PV-induced alarmin (HMGB1) secretion with other (ICD-inducing) chemotherapeutic regimens warrants the consideration of PV inclusion into various multimodal anti-PDAC treatment protocols. The therapeutic promise of H-1PV administration in gemcitabine-treated pancreatic cancer patients is further supported by several reports in the literature showing that, unlike most nucleoside analogues, gemcitabine is lacking immunosuppressive properties. On the contrary, gemcitabine may be beneficial not only to the cytocidal but also to the proimmune outcome of H-1PV infection.
Based on favorable preclinical data hinting at the potentiation of OV-induced antitumor effects in the presence of gemcitabine, a clinical trial, ParvOryx02 (NCT02653313), was designed and conducted with the aim to provide a clinical proof-of-principle of the safety (and efficacy) of H-1PV+gemcitabine co-treatment. Patients with inoperable metastatic (at least one hepatic metastasis) pancreatic cancer were treated with H-1PV. The virus was first administered intravenously (40% of the total virus dose on four consecutive days), and the remaining virus dose was then given intrametastatically as single hepatic injection, followed by gemcitabine treatment. Partial response and extended overall survival were observed in two out of seven trial patients, and immunological signatures most likely contributed to this improved outcome. The ParvOryx02 study therefore provided the first clinical indication that immune mechanisms underlie PV-mediated tumor suppression [5].
H-1PV + Histone Deacetylase Inhibitors (Valproic Acid)
Preclinical proof-of-concept was also obtained for another treatment combining H-1PV with the histone deacetylase (HDAC) inhibitor (HDACi) valproic acid (VPA). HDACis hold significant promise in cancer therapy, due to their ability to cause malignant cell growth inhibition, re-differentiation and death. Most interestingly, HDAC inhibition was also found to potentiate the oncotoxicity of various OVs, including vesicular stomatitis, herpes-, adeno- and parvo- viruses (for a review, see [6]). The synergism between HDACi and H-1PV was first demonstrated by Li et al., who conducted preclinical testing of this combination in cervical carcinoma and PDAC models. VPA proved to synergize with H-1PV in inducing DNA damage, oxidative stress and death in PDAC-derived cell lines. This cooperation was traced back, at least in part, to the ability of VPA to stimulate the acetylation and, in consequence, the oncotoxic activity of the viral protein NS1. Interestingly, VPA-induced hyperacetylation of NS1 was also associated with enhanced H-1PV DNA replication and viral gene transcription, ultimately boosting virus multiplication in tumor cells. The VPA-dependent increase in both H-1PV intrinsic oncotoxicity and multiplication was reflected in the potentiation of tumor suppression in animal models. In order to establish a clinically relevant animal model of PDAC, patient-derived material was xenotransplanted in nonobese diabetic/severe combined immunodeficiency disease (NOD/SCID) mice. Alternatively, the human AsPC-1 cell line was implanted into nude rats. Tumors were subjected to mono versus combinatorial treatment and tumor growth parameters were comparatively evaluated. In line with the in vitro observations, H-1PV+VPA administration resulted in enhanced NS1 and H-1PV intratumoral accumulation, correlating with an increase in oxidative stress and subsequent apoptosis in co-treated tumors. The combination achieved complete AsPC-1 tumor eradication. Patient-derived xenografts were also responsive, yet to a somewhat lesser extent, probably due to the characteristic PDAC intratumoral heterogeneity and prominent presence of stroma.
H-1PV + Proinflammatory Cytokines (Interferon-Gamma)
Another combination with substantial potential for clinical development relies on the mutual complementation of H-1PV- and IFN-γ-mediated immune stimulation. It was shown that IFN-γ improves the vaccination potential of the virus and diminishes the development of peritoneal carcinomatosis in preclinical PDAC models. Concomitant intraperitoneal administration of both H-1PV and IFN-γ in these models led to extended animal survival correlating with enhanced peritoneal macrophage and splenocyte responses against tumor cells.
Parvovirus-Based Combinatorial Immunotherapy against Glioblastoma
Based on the so far unmet need for novel, more efficient treatments, glioblastoma multiforme (GBM) was among the preclinical tumor models most extensively studied in our laboratory. H-1PV capacity for selectively killing glioma cells through cytosolic activation of lysosomal proteases was first demonstrated in vitro. These results were validated in animal models, namely in immunocompetent rats bearing orthotopic autologous RG-2 tumors and in immunodeficient rats bearing xenotransplanted human U87 gliomas. In these models, tumor regression after local, intravenous or intranasal virus administration was observed. H-1PV treatment was not associated with any significant off-target toxicities; accordingly, virus transcription and NS1 protein accumulation could be detected in regressing tumor remnants and not in the surrounding normal tissues. Interestingly, the therapeutic effect was potentiated in the presence of an intact host immune system. T cell depletion impaired H-1PV-induced glioma suppression; conversely, the presence of T cell only, in the absence of PV treatment, was not sufficient to inhibit tumor growth. These preclinical observations provided the first hints of host T cell response involvement in PV-mediated glioma regression, hence the rationale for the development of PV-based immunotherapies against glioblastoma.
Pursuant to the above-described preclinical findings, the ParvOryx01 trial (NCT01301430) in recurrent glioblastoma patients delivered the first clinical proof-of-concept for tumor-infiltrating lymphocytes (TILs) playing substantial role in H-1PV-mediated immunomodulation of GBM TME. Although ParvOryx01 primary objectives were to determine virus safety, tolerability, pharmacokinetics, shedding and maximum tolerated dose, the analysis of post-virus-treatment resected tumor tissues revealed the presence of prominent immune cell infiltrates [7]. These infiltrates were comprised of CD45+CD3+CD4+ and CD45+CD3+CD8+ TILs. The latter contained both perforin and granzyme B-positive secretory granules, which is indicative of CTL cytolytic activity. TILs proved, in addition, to be CD25 (IL2 receptor alpha chain)-positive. Only a minor fraction of these cells expressed FOXP3, indicating the scarcity of Treg cells within the intratumoral immune infiltrates. Intratumoral production of proinflammatory cytokines (IFN-γ, IL-2) was also detected, together with inducible nitric oxide synthase (iNOS) expression in CD68+ tumor-associated microglia/macrophage cells. Interestingly, tumor cells expressed the CD40 ligand (CD40L), a positive prognostic factor in glioblastoma. Co-expression of CD40L and CD40, considered as a negative prognostic factor, was not seen. Taken together, these first clinical findings indicated that H-1PV has the capacity to exert immunostimulating effects on glioblastoma TME. This makes the virus a worthwhile partner in therapeutic combinations, which aim at warming up the intrinsically immunosuppressive and immune-evasive environment of brain tumors.
H-1PV + Ionizing Radiation
We have previously shown that radiotherapy, one of the conventional first-line treatments in glioblastoma patients, sensitizes low-passage glioma cultures to H-1PV oncolysis. Pre-irradiation increases the susceptibility of these cells to virus infection. Interestingly, H-1PV achieves killing both radiation-sensitive and resistant glioma cells. Apart from triggering enhanced tumor cytolysis, the irradiation followed by H-1PV treatment holds, in addition, the potential—although not yet validated in animal models—of acting as combinatorial immunotherapy. Indeed, although irradiation was long regarded as a local anticancer therapy, the first reports on radiotherapy interactions with the host immune system can be traced back to the 1970s of the last century. In 1979, Slone et al. were the first to report that the radiation dose required to control 50% of mouse fibrosarcomas was twice as high in immunocompromised animals as in immunocompetent hosts. Furthermore, tumor regression at sites distant to radiation fields, the so-called abscopal effect, has been systematically observed. Radiation-triggered immunomodulation encompasses, among other effects, ICD induction, T and NK cell activation and MDSC suppression. These observations prompted the development of various combination therapy regimens based on radiation and other immunomodulating agents, including OVs (e.g., adeno-, herpes simplex-, measles- and vaccinia- viruses) against glioma.
H-1PV + Tumor Angiogenesis Inhibitors (Bevacizumab)
Another promising approach is the combination of H-1PV with bevacizumab (Avastin®). This co-treatment was evaluated in a series of compassionate virus uses in recurrent glioblastoma patients. Bevacizumab is an anti-vascular endothelial growth factor (VEGF)-A receptor monoclonal antibody available in Europe since 2005 for the treatment of breast, lung, kidney, colon, ovarian and endometrial carcinomas. In 2009, bevacizumab was approved by the Food and Drug Administration (FDA) for application in glioblastoma patients. While achieving a steroid-sparing effect and alleviation of edema, bevacizumab monotherapy has, however, not demonstrated significant survival benefits. On the other hand, scientists and clinicians have gathered an extensive—and yet to grow—knowledge of bevacizumab’s mode of action. In particular, bevacizumab was found to exert immunomodulating activity by counteracting VEGF-induced negative effects on dendritic cell (DC) maturation, antigen presentation and lymphocytic trafficking. These bevacizumab properties, together with the ParvOryx01 trial experience showing H-1PV treatment-associated immunogenic changes in glioblastoma TME, have opened up prospects for novel anti-glioma combinatorial immunotherapy development, namely, H-1PV+bevacizumab. A compassionate use proof-of-concept program was conducted in five GBM patients, who developed a second or third recurrence after being treated in the ParvOryx01 trial. The patients underwent tumor resection, followed by local H-1PV administration and bevacizumab. The mean survival after treatment was extended to 15.4 months. Moreover, in three out of the five patients, striking remission of the recurrence was observed, providing first clinical hints of synergistic glioblastoma suppression through parvoviro-immunotherapy [8].
H-1PV + PD-1 Immune Checkpoint Inhibitors (Nivolumab)
Checkpoint blockade, a strategy which aims at overcoming immune system tolerance towards the tumor through the release from negative regulators of immune activation (immune checkpoints), is presently at the leading edge of cancer immunotherapy. Although efficient in controlling various other solid tumors, immune checkpoint inhibitors (ICIs) frequently fail to achieve a significant response in glioblastoma patients. Several preclinical studies and clinical trials have therefore been initiated, in order to determine the optimal ICI-based combinations and redefine the future standards of care for this deadly disease.
First clinical hints of improved antitumor effects of H-1PV virotherapy upon combination with checkpoint blockade were obtained through compassionate virus uses. A series of three patients with rapidly progressing recurrent glioblastoma were treated with H-1PV (two were irradiated prior to virus administration), followed by bevacizumab and the programmed cell death protein 1 (PD-1) inhibitor nivolumab. In addition, all patients received the HDACi VPA. This innovative PV-based multimodal strategy led to radiologically confirmed tumor regression accompanied by clinical improvement in all subjects 4 to 8 weeks after virus injection [9]. An objective tumor response was also seen in another group of primary or recurrent glioblastoma patients, who received H-1PV in combination with bevacizumab and checkpoint blockade. Complete to partial tumor remission was documented in 78% of the cases, which is a significantly higher response rate than the one reported in the literature for bevacizumab- and ICI-based monotherapies [10].
Altogether, the above data provide a strong impetus for further clinical development of H-1PV combinations with radiation and/or immunomodulators (in particular bevacizumab and ICIs) in the fight against glioblastoma.
References
- Bretscher, C., Marchini, A.; H-1 parvovirus as a cancer-killing agent: Past, present, and future. Viruses 2019, 11, 562, https://doi.org/10.3390/v11060562.
- Rommelaere, J. et al.. Viral Therapy of Human Cancers; Sinkovics, J.G., Horvath, J.C., Eds.; Marcel Dekker : New York, NY, USA, 2005; pp. 627–675.
- Angelova, A., Rommelaere, J.; Immune System Stimulation by Oncolytic Rodent Protoparvoviruses. Viruses 2019, 11, 415, https://doi.org/10.3390/v11050415.
- Angelova, A. et al.; Improvement of Gemcitabine-Based Therapy of Pancreatic Carcinoma by Means of Oncolytic Parvovirus H-1PV. Clin. Cancer Res 2009, 15, 511–519, 10.1158/1078-0432.CCR-08-1088.
- ParvOryx02: A Phase II Trial of Intravenous and Intratumoral Administration of H-1 Parvovirus in Patients with Metastatic Pancreatic Cancer . oryx-medicine.com. Retrieved 2021-1-29
- Marchini, A. et al.; Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9, https://doi.org/10.3390/v8010009.
- Geletneky, K. et al.; Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017, 25, 2620–2634, https://doi.org/10.1016/j.ymthe.2017.08.016.
- Geletneky, K. et al.; Favorable response of patients with glioblastoma at second or third recurrence to repeated injection of oncolytic parvovirus H-1 in combination with bevacicumab. Neuro Oncol 2015, 17, v11, https://doi.org/10.1093/neuonc/nov205.07.
- Geletneky, K. et al.; First clinical observation of improved anti-tumor effects of viro-immunotherapy with oncolytic parvovirus H-1 in combination with PD-1 checkpoint blockade and bevacicumab in patients with recurrent glioblastoma. Neuro Oncol 2016, 18, vi24, https://doi.org/10.1093/neuonc/now212.094.
- Geletneky, K. et al.; High rate of objective anti-tumor response in 9 patients with glioblastoma after viro-immunotherapy with oncolytic parvovirus H-1 in combination with bevacicumab and PD-1 checkpoint blockade. Neuro Oncol 2018, 20, vi10, https://doi.org/10.1093/neuonc/noy148.035.