The AMP-activated protein kinase (AMPK) is the central component of a signaling pathway that is conserved in essentially all eukaryotes, the exceptions being a few parasites (e.g., Plasmodium falciparum, the causative agent of malaria) that spend most of their life cycle living inside other eukaryotic cells, in which case the host cell provides AMPK and the parasite may therefore have been able to dispense with it. AMPK is activated by various stresses that act via both classical (canonical) and non-classical (non-canonical) pathways. Pharmacological activation of AMPK can also be achieved via a range of mechanisms. Here, we enumerate the different classes of AMPK activators and describe their mechanisms of action.
- Pharmacological Activation of AMPK
1. Introduction
The idea that activation of AMPK might be useful in treatment of disorders of whole body energy balance such as obesity and Type 2 diabetes, which appears to have been first proposed in 1999
[1], led to a drive towards the development of AMPK activators by pharmaceutical and biotechnology companies. These campaigns have now given rise to several different classes of activator that are discussed below. There has been much less emphasis on the development of inhibitors, although (as mentioned in the previous paragraph) these may now be regarded as good candidates for the treatment of some cancers. The limited current progress in this direction is discussed below.
, led to a drive towards the development of AMPK activators by pharmaceutical and biotechnology companies. These campaigns have now given rise to several different classes of activator that are discussed below. There has been much less emphasis on the development of inhibitors, although (as mentioned in the previous paragraph) these may now be regarded as good candidates for the treatment of some cancers. The limited current progress in this direction is discussed below.
2. AMPK Activators: Pro-Drugs That Are Converted to AMP Analogues
2. AMPK Activators: Pro-Drugs That Are Converted to AMP Analogues
The first pharmacological agent shown to activate AMPK was 5-aminoimidazole-4-carboxamide ribonucleoside
, which those in the AMPK field often refer to as AICAR. Unfortunately, this acronym can cause confusion because researchers in the field of nucleotide metabolism use the same term to refer to the monophosphorylated nucleotide, also sometimes known as ZMP; to avoid this, we will refer to the nucleoside as AICA riboside and to the nucleotide as ZMP. The incubation of cells in vitro (or injection of animals in vivo) with AICA riboside usually leads to activation of AMPK because the riboside is rapidly taken up into cells by adenosine transporters
[5]
and rapidly converted to ZMP by adenosine kinase
[6]
(
). ZMP, which is an AMP analogue, then mimics the multiple effects of AMP on the AMPK system, including allosteric activation and protection against Thr172 dephosphorylation. Although ZMP is around 50-fold less potent in these effects than AMP itself, AICA riboside is nevertheless effective at activating AMPK in many cells because ZMP accumulates to millimolar concentrations in their cytoplasm
[4]
. AICA riboside was historically used as a means of artificial activation of AMPK in vitro and in vivo, and is still sometimes used today. However, it is perhaps not surprising that millimolar levels of any ligand can have off-target effects. For example, ZMP mimics the effects of AMP on other AMP-sensitive enzymes, including the glycogenolytic enzyme glycogen phosphorylase in cardiac muscle
[7]
and the gluconeogenic enzyme fructose-1,6-bisphosphatase in the liver
[8]
. Indeed, some of the effects of AICA riboside on hepatic glucose production, which were originally ascribed to AMPK activation, can now be put down to inhibition of fructose-1,6-bisphosphatase by ZMP
.
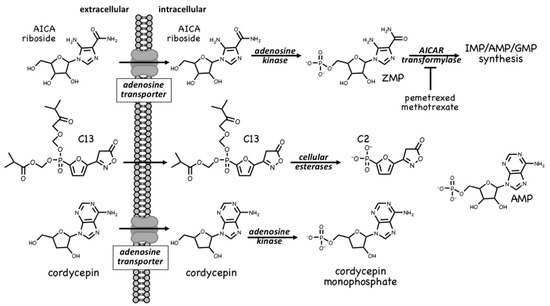
ZMP is, in fact, a naturally-occurring metabolite in the pathway of de novo purine nucleotide synthesis, being converted in two further steps to IMP, the common precursor for AMP and GMP (Figure 1). These two steps are catalysed by the enzymes AICAR transformylase and IMP cyclohydrolase, which are carried on a single polypeptide chain encoded by the ATIC gene. The rapid metabolism of ZMP to IMP explains why AMPK is not activated by AICA riboside in some cell types, especially in proliferating cells that have a high capacity for de novo nucleotide biosynthesis. AICAR transformylase (note that AICAR here refers to the nucleotide, i.e., ZMP) uses N10-formyltetrahydrofolate to add an aldehyde group to what is then converted by IMP cyclohydrolase into the 6-membered ring of IMP. Because of this, AICAR transformylase is inhibited by folate analogues such as methotrexate and pemetrexed, which are used in the treatment of some cancers and autoinflammatory disorders. While the primary target of these antifolate drugs is thought to be thymidylate synthase and hence DNA synthesis, AICAR transformylase may be a secondary target. Thus, pemetrexed has been shown to activate AMPK in leukaemia cells due to inhibition of AICAR transformylase and consequent accumulation of its substrate ZMP[11], while in HEK-293 cells, where AICA riboside has little effect on its own because of rapid metabolism of ZMP by AICAR transformylase, the ribonucleoside does activate AMPK dramatically in the presence of the AICAR transformylase inhibitor, methotrexate [12]. Whether these effects on AMPK explain any of the anti-cancer actions of antifolate drugs remains unclear at present.
A much more selective AMPK activator that works through a different pro-drug mechanism is C13, which contains a phosphonate group esterified on two of its oxygen atoms (Figure 1). This modification makes C13 cell-permeable but, once inside cells, it is converted by intracellular esterases into C2, a phosphonate analogue of AMP that, remarkably, is at least two orders of magnitude more potent as an allosteric activator of AMPK than AMP itself [13]. An explanation for this surprising finding is that although its binding site on the AMPK-γ subunit overlaps with that of AMP, the two nucleotides bind in different orientations[14]. Another notable feature of C2 is that it is almost completely selective for AMPK complexes containing the α1 isoform, with little or no effect on α2 complexes. One major advantage of the use of C13, compared with AICA riboside, is that its active metabolite C2 does not affect the regulation of other AMP-sensitive enzymes such as glycogen phosphorylase and fructose-1,6-biphosphatase [15].
A final compound that activates AMPK by a pro-drug mechanism related to that of AICA riboside is cordycepin, also known as 3′-deoxyadenosine (Figure 1). Cordycepin is an adenosine analog derived from species of the genus Cordyceps, which are parasitic fungi (highly prized in traditional Chinese medicine) that infect insect larvae such as caterpillars and (rather gruesomely!) consume them from within [16]. Cordycepin is an adenosine analogue that is taken up into cells and converted by cellular metabolism into mono-, di- and tri-phosphates. Since it lacks a hydroxyl group at the 3′-position on the ribose ring, if cordycepin is incorporated into RNA it will cause chain termination. Poly(A) polymerases, which add the poly(A) tails to mRNA, seem to accept cordycepin in place of adenosine particularly readily, so that treatment of cells with cordycepin reduces the lengths of the poly(A) tails, and hence the stability, of many mRNAs. This may be the main reason why cordycepin has cytotoxic effects to reduce cell viability. However, two groups reported in 2010 that cordycepin treatment also activated AMPK[17][18]. Our group has provided evidence that this occurs because cordycepin is taken up into cells via adenosine transporters, and is converted by adenosine kinase into cordycepin monophosphate (Figure 1), which then mimics the multiple effects of AMP to activate AMPK[6]. Cells lacking AMPK are also more sensitive to the cytotoxic effects of cordycepin, suggesting that the ability of the compound to activate AMPK normally ameliorates its cytotoxic effects.
3. AMPK Activators: Indirect Activation via Inhibition of ATP Synthesis
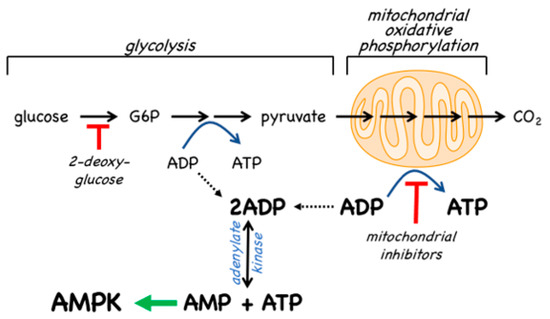
Since the inhibition of ATP synthesis will cause increases in the ADP:ATP ratio that are amplified by adenylate kinases into even larger increases in the AMP:ATP ratio, this will inevitably also cause a secondary, indirect activation of AMPK (Figure 2). For example, in cells that are partly reliant on glycolysis for ATP production, addition of the glycolytic inhibitor 2-deoxyglucose causes rapid AMPK activation[19]. However, the major generator of ATP in most animal cells is not glycolysis but mitochondrial oxidative metabolism, and many pharmacological agents that activate AMPK do so indirectly by inhibiting the latter (Figure 2). In particular, Complex I of the respiratory chain (NADH–ubiquinone oxidoreductase) is a remarkable membrane-bound multiprotein machine with 14 core subunits and around 30 accessory subunits[20]. It is perhaps not surprising that many xenobiotic compounds that are hydrophobic in nature should find binding sites within this complex, where they cause its inhibition, with consequent downstream activation of AMPK. Many compounds known to activate AMPK by this mechanism are secondary products of plants and may be produced by the plant to poison herbivorous insects and other animals, and thus deter grazing. These compounds are often stored in the cell wall or vacuole of the producing plant cell, where they will not come into contact with the mitochondria of the plant itself [21]. An example of a natural plant product that activates AMPK is galegine[22], which is derived from the plant Galega officinalis. The latter (also known as Goat’s Rue, presumably because it is poisonous to goats!) was recommended as a herbal remedy in a medical text [23] published as long ago as 1640 by John Parkinson, the court physician to King James I of England. Galegine itself, an isoprenyl guanidine, turned out to be too toxic for human use, but in the 20th century various biguanide derivatives, including metformin and phenformin, were synthesized. In the second half of the century these became mainstays of the treatment of Type 2 diabetes, although phenformin was withdrawn in the late 1970s because in rare cases it was associated with a life-threatening lactic acidosis. In 2000, two groups reported that metformin and phenformin were inhibitors of Complex I of the respiratory chain[24][25] (providing, incidentally, an explanation for the lactic acidosis associated with phenformin use), and the following year it was reported that metformin activated AMPK[26]. Although there have been various proposals to explain the mechanism of action of biguanides other than by inhibition of Complex I and other than by AMPK activation[27][28], there is general agreement that these drugs do indirectly activate AMPK.
Traditional herbal medicines are a particularly rich source of natural plant products that activate AMPK—a 2016 review listed at least 100 such compounds [29], and more are still being reported in multiple papers every week. In most cases, the mechanism(s) by which these compounds activate AMPK have not been established. However, berberine [30] and arctigenin [31] have been shown to inhibit Complex I of the respiratory chain while galegine, quercetin, resveratrol, berbamine, and mangiferin have all been suggested to activate AMPK indirectly by altering cellular adenine nucleotide ratios [32][33][34].
4. AMPK Activators: Drugs and Metabolites Binding at the AdaM Site
Several pharmaceutical companies have isolated direct activators of AMPK from compound libraries via high-throughput screens that searched for allosteric activators of the purified kinase (Figure 3). First-in-class was A-769662, which had favourable effects on several metabolic parameters when injected into genetically obese (ob/ob) mice, but had poor oral availability[35]. Mechanistic studies suggested that although A-769662 binding had similar effects to AMP (causing both allosteric activation and inhibition of Thr172 dephosphorylation) it bound at different site(s), and the interactions appeared to involve the β subunit because mutations in the latter abolished activation by A-769662 but not AMP[36][37]. Also supporting a role for the β subunit were findings that A-769662 only activated AMPK complexes containing β1 and not the β2 isoform [38]. Additional screens led to the development of more potent activators in this class, such as MT 63-78[39], PF-06409577 [40], PF-249 [41], PF-739 [42], and MK-8722[43] (Figure 3). Several of these compounds have much better oral availability than A-769662 and some, e.g., PF-739 [42], and MK-8722[43], are termed “pan-β” activators in that they activate β2-containing complexes almost as well as β1 complexes. Because β2 is the major β subunit isoform expressed in skeletal muscle, both PF-739 and MK-8722 are effective in promoting glucose uptake by skeletal muscle, and this appears to be why they are more efficacious in ameliorating metabolic defects in animal models of obesity and Type 2 diabetes than β1-selective activators, such as A-769662 or PF-249[43][42].
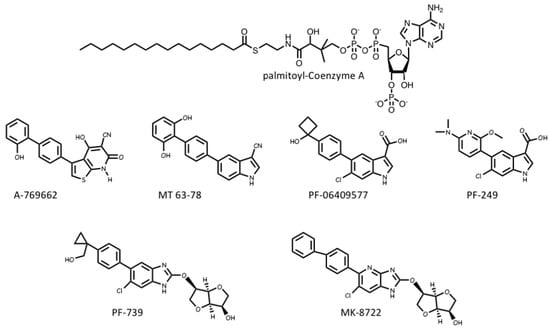
The crystallization of human AMPK complexes with 991 (a lead compound identified during development of MK-8722), or A-769662 or its halogenated analogues, finally definitively identified the binding site for these compounds. They bind in a deep hydrophobic cleft located between the β-CBM (the opposite surface to the glycogen-binding site) and the N-lobe of the α-KD (the opposite surface to the MgATP2--binding site). Because this site lies between the α and β subunits of the complex, it is unique to AMPK, and therefore a potentially specific target site for therapeutics. A curious feature of this site is that essentially all of the compounds that were initially found to activate AMPK by binding to it were derived from libraries of synthetic small molecules, rather than natural products. However, most researchers in the field believe these compounds must be mimicking a metabolite that occurs naturally in mammalian cells, hence its designation as the allosteric drug and metabolite (ADaM) site[44]. One natural product that does bind the ADaM site is salicylate, an active metabolite of the drug acetyl salicylic acid (ASA or aspirin). Salicylate, or derivatives such as salicin and methyl salicylate, are used by higher plants as hormones that signal infection by pathogens [45]. Salicylate was found to activate AMPK in a manner competitive with A-769662[46], and an iodinated analogue of salicylate bound to the same site as A-769662 [47]. However, salicylate would not occur naturally in animals, except perhaps if plants suffering from a pathogen infection had been ingested.
Recently, it has been suggested by Pinkosky et al.[48] that the natural ligands that activate AMPK by binding to the ADaM site may be long-chain fatty acyl-CoA esters (LCFA-CoAs). Ironically, this brings the story of regulation of AMPK full circle, as one of the first papers to define AMPK and demonstrate its sensitivity to AMP also noted that its activation by an upstream kinase (at that time unidentified) was enhanced by palmitoyl-CoA[49]. Subsequently, activation of AMPK by LCFAs was found in perfused rat heart in vitro and in rat liver in vivo [50][51], and it was also reported that CoA esters of LCFAs or LCFA analogues enhanced phosphorylation of AMPK by LKB1[52][53].
In this latest paper[48], the structural basis for the regulation of AMPK by LCFAs has been addressed. It was found that micromolar concentrations of LCFA-CoAs containing saturated or mono-unsaturated fatty acids of 12 carbons or more activated AMPK, whereas the corresponding free acids or carnitine esters did not. Moreover, like A-769662, palmitoyl-CoA (C16, saturated) only activated β1-containing complexes, and β-CBM mutations that perturbed the ADaM site, but not γ subunit mutations affecting the nucleotide-binding sites, eliminated or reduced palmitoyl-CoA-induced AMPK activation. Note that, in cells subject to glucose starvation, LCFA oxidation in mitochondria would represent a crucial alternative source of ATP. The activation of AMPK by LCFA-CoAs, derived either from external sources or from breakdown of stored triglycerides, would therefore represent a type of “feed-forward” activation that would trigger enhanced LCFA oxidation via phosphorylation of acetyl-CoA carboxylases (ACC1/ACC2), with consequent relief of malonyl-CoA-mediated inhibition of LCFA uptake into mitochondria by the carnitine:palmitoyl-CoA transferase system [54]. Consistent with this, Pinkosky et al. [48] found that incubation of mouse hepatocytes with LCFAs promoted phosphorylation of ACC1/ACC2 at the equivalent AMPK sites (S79/S221), while oral administration of a triglyceride and phospholipid emulsion in vivo promoted fat oxidation in wild type mice, but not in knock-in mice in which both S79 and S221 had been mutated to alanine. If this intriguing model is correct, AMPK represents not only a sensor of cellular energy status and an indirect sensor of the availability of glucose (and perhaps also glycogen), but also a direct sensor of fatty acid availability.
5. AMPK Inhibitors
Perhaps because the suggestion that inhibitors of AMPK might be useful in the treatment of cancer has only been made fairly recently, there has been much less progress in their development than for AMPK activators. In 2001, a screen identified compound C as an inhibitor of AMPK that appeared to bind to the MgATP2--binding site in the kinase domain[26]. It did not inhibit any of a panel of 5 other protein kinases[26] and, as a result, is still often claimed to be a “highly selective” inhibitor of AMPK. However, this is not the case; one early indication that compound C has multiple targets was that it is identical with dorsomorphin, which was identified in an independent screen for compounds that perturb dorsoventral axis formation in zebrafish and is an inhibitor of BMP (bone morphogenetic protein) signalling acting independently of AMPK[55]. Moreover, in a screen of around 70 protein kinases, Bain et al.[56] reported that at least ten were inhibited by compound C to a greater extent than AMPK, while in recent screens of more than 100 protein kinases available via the MRC Kinase Profiling Inhibitor Database[57], up to 30 were inhibited to a greater extent than AMPK. As stated by Bain et al. [56]: “the use of this compound to identify potential functions of AMPK is not recommended”. The field therefore awaits the development of a truly specific inhibitor and, despite the widespread use of compound C as an AMPK inhibitor in the literature, currently the only way to be sure that AMPK is involved in a cellular process is to use genetic approaches such as gene knockouts.
References
- Winder, W.W.; Hardie, D.G. The AMP-activated protein kinase, a metabolic master switch: Possible roles in Type 2 diabetes. Am. J. Physiol. 1999, 277, E1–E10.
- Henin, N.; Vincent, M.F.; Gruber, H.E.; Van den Berghe, G. Inhibition of fatty acid and cholesterol synthesis by stimulation of AMP-activated protein kinase. FASEB J. 1995, 9, 541–546.
- Sullivan, J.E.; Brocklehurst, K.J.; Marley, A.E.; Carey, F.; Carling, D.; Beri, R.K. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994, 353, 33–36.
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-carboxamide ribonucleoside: A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565.
- Gadalla, A.E.; Pearson, T.; Currie, A.J.; Dale, N.; Hawley, S.A.; Sheehan, M.; Hirst, W.; Michel, A.D.; Randall, A.; Hardie, D.G.; et al. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J. Neurochem. 2004, 88, 1272–1282.
- Hawley, S.A.; Ross, F.A.; Russell, F.M.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanism of activation of AMPK by cordycepin. Cell Chem. Biol. 2020, 27, 214–222.
- Longnus, S.L.; Wambolt, R.B.; Parsons, H.L.; Brownsey, R.W.; Allard, M.F. 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol. 2003, 284, R936–R944.
- Vincent, M.F.; Marangos, P.J.; Gruber, H.E.; Van den Berghe, G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes 1991, 40, 1259–1266.
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010, 120, 2355–2369.
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406.
- Racanelli, A.C.; Rothbart, S.B.; Heyer, C.L.; Moran, R.G. Therapeutics by cytotoxic metabolite accumulation: Pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009, 69, 5467–5474.
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369.
- Gomez-Galeno, J.E.; Dang, Q.; Nguyen, T.H.; Boyer, S.H.; Grote, M.P.; Sun, Z.; Chen, M.; Craigo, W.A.; van Poelje, P.D.; MacKenna, D.A.; et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med. Chem. Lett. 2010, 1, 478–482.
- Langendorf, C.G.; Ngoei, K.R.; Scott, J.W.; Ling, N.X.; Issa, S.M.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912.
- Hunter, R.W.; Foretz, M.; Bultot, L.; Fullerton, M.D.; Deak, M.; Ross, F.A.; Hawley, S.A.; Shpiro, N.; Viollet, B.; Barron, D.; et al. Mechanism of action of compound-13: An alpha1-selective small molecule activator of AMPK. Chem. Biol. 2014, 21, 866–879.
- Tuli, H.S.; Sharma, A.K.; Sandhu, S.S.; Kashyap, D. Cordycepin: A bioactive metabolite with therapeutic potential. Life Sci. 2013, 93, 863–869.
- Wong, Y.Y.; Moon, A.; Duffin, R.; Barthet-Barateig, A.; Meijer, H.A.; Clemens, M.J.; de Moor, C.H. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J. Biol. Chem. 2010, 285, 2610–2621.
- Guo, P.; Kai, Q.; Gao, J.; Lian, Z.Q.; Wu, C.M.; Wu, C.A.; Zhu, H.B. Cordycepin prevents hyperlipidemia in hamsters fed a high-fat diet via activation of AMP-activated protein kinase. J. Pharmacol. Sci. 2010, 113, 395–403.
- Tsou, P.; Zheng, B.; Hsu, C.H.; Sasaki, A.T.; Cantley, L.C. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011, 13, 476–486.
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388.
- Shitan, N. Secondary metabolites in plants: Transport and self-tolerance mechanisms. Biosci. Biotechnol. Biochem. 2016, 80, 1283–1293.
- Mooney, M.H.; Fogarty, S.; Stevenson, C.; Gallagher, A.M.; Palit, P.; Hawley, S.A.; Hardie, D.G.; Coxon, G.D.; Waigh, R.D.; Tate, R.J.; et al. Mechanisms underlying the metabolic actions of galegine that contribute to weight loss in mice. Br. J. Pharmacol. 2008, 153, 1669–1677.
- Parkinson, J. Galega, Goat’s Rue. In Theatricum Botanicum; Thomas Cotes: London, UK, 1640; pp 417–418.
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614.
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174.
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585.
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin activates AMPK through the lysosomal pathway. Cell Metab. 2016, 24, 521–522.
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of liver AMPK with PF-06409577 corrects NAFLD and lowers cholesterol in rodent and primate preclinical models. EBioMedicine 2018, 31, 122–132.
- Turner, N.; Li, J.Y.; Gosby, A.; To, S.W.; Cheng, Z.; Miyoshi, H.; Taketo, M.M.; Cooney, G.J.; Kraegen, E.W.; James, D.E.; et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008, 57, 1414–1418.
- Huang, S.L.; Yu, R.T.; Gong, J.; Feng, Y.; Dai, Y.L.; Hu, F.; Hu, Y.H.; Tao, Y.D.; Leng, Y. Arctigenin, a natural compound, activates AMP-activated protein kinase via inhibition of mitochondria complex I and ameliorates metabolic disorders in ob/ob mice. Diabetologia 2012, 55, 1469–1481.
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565.
- Sharma, A.; Anand, S.K.; Singh, N.; Dwivedi, U.N.; Kakkar, P. Berbamine induced AMPK activation regulates mTOR/SREBP-1c axis and Nrf2/ARE pathway to allay lipid accumulation and oxidative stress in steatotic HepG2 cells. Eur. J. Pharmacol. 2020, 882, 173244.
- Li, J.; Liu, M.; Yu, H.; Wang, W.; Han, L.; Chen, Q.; Ruan, J.; Wen, S.; Zhang, Y.; Wang, T. Mangiferin improves hepatic lipid metabolism mainly through its metabolite-norathyriol by modulating SIRT-1/AMPK/SREBP-1c signaling. Front. Pharmacol. 2018, 9, 201.
- Cool, B.; Zinker, B.; Chiou, W.; Kifle, L.; Cao, N.; Perham, M.; Dickinson, R.; Adler, A.; Gagne, G.; Iyengar, R.; et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006, 3, 403–416.
- Goransson, O.; McBride, A.; Hawley, S.A.; Ross, F.A.; Shpiro, N.; Foretz, M.; Viollet, B.; Hardie, D.G.; Sakamoto, K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J. Biol. Chem. 2007, 282, 32549–32560.
- Sanders, M.J.; Ali, Z.S.; Hegarty, B.D.; Heath, R.; Snowden, M.A.; Carling, D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J. Biol. Chem. 2007, 282, 32539–32548.
- Scott, J.W.; van Denderen, B.J.; Jorgensen, S.B.; Honeyman, J.E.; Steinberg, G.R.; Oakhill, J.S.; Iseli, T.J.; Koay, A.; Gooley, P.R.; Stapleton, D.; et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem. Biol. 2008, 15, 1220–1230.
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538.
- Hardie, D.G. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19.
- Salatto, C.T.; Miller, R.A.; Cameron, K.O.; Cokorinos, E.; Reyes, A.; Ward, J.; Calabrese, M.; Kurumbail, R.; Rajamohan, F.; Kalgutkar, A.S.; et al. Selective activation of AMPK b1-containing isoforms improves kidney function in a rat model of diabetic nephropathy. J. Pharmacol. Exp. Ther. 2017, 361, 303–311.
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjobsted, R.; Jorgensen, N.O.; Tran, J.L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017, 25, 1147–1159.
- Myers, R.W.; Guan, H.P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Trotter, D.G.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511.
- Langendorf, C.G.; Kemp, B.E. Choreography of AMPK activation. Cell Res. 2015, 25, 5–6.
- Reymond, P.; Farmer, E.E. Jasmonate and salicylate as global signals for defense gene expression. Curr. Opin. Plant Biol. 1998, 1, 404–411.
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922.
- Calabrese, M.F.; Rajamohan, F.; Harris, M.S.; Caspers, N.L.; Magyar, R.; Withka, J.M.; Wang, H.; Borzilleri, K.A.; Sahasrabudhe, P.V.; Hoth, L.R.; et al. Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 2014, 22, 1161–1172.
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK beta1 isoforms. Nat. Metab. 2020, 2, 873–881.
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 223, 217–222.
- Clark, H.; Carling, D.; Saggerson, D. Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids. Eur. J. Biochem. 2004, 271, 2215–2224.
- Suchankova, G.; Tekle, M.; Saha, A.K.; Ruderman, N.B.; Clarke, S.D.; Gettys, T.W. Dietary polyunsaturated fatty acids enhance hepatic AMP-activated protein kinase activity in rats. Biochem. Biophys. Res. Commun. 2005, 326, 851–858.
- Watt, M.J.; Steinberg, G.R.; Chen, Z.P.; Kemp, B.E.; Febbraio, M.A. Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J. Physiol. 2006, 574 Pt 1, 139–147.
- Za’tara, G.; Bar-Tana, J.; Kalderon, B.; Suter, M.; Morad, E.; Samovski, D.; Neumann, D.; Hertz, R. AMPK activation by long chain fatty acyl analogs. Biochem. Pharmacol. 2008, 76, 1263–1275.
- Hardie, D.G.; Carling, D. The AMP-activated protein kinase: Fuel gauge of the mammalian cell? Eur. J. Biochem. 1997, 246, 259–273.
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41.
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315.
- MRC Kinase Profiling Inhibitor Database. Available online: http://www.kinase-screen.mrc.ac.uk/kinase-inhibitors (accessed on 30 September 2020).