Asthma is a complex and multifactorial respiratory disease with a high prevalence in the pediatric population. Variation in treatment response to asthma therapies has been described among patients, and difficult-to-treat asthma carries both high healthcare and socioeconomic burden to the patients and society. Omic studies can be used to discover the molecular mechanisms underlying asthma susceptibility and treatment response, contributing to a better knowledge and definition of asthma pathogenesis and therefore, to the development of precision medicine. This entry aims to summarize the recent findings of omic studies of treatment response in childhood asthma. Between 2018-2019 a total of 13 omic studies has been performed involving genomics, epigenomics, transcriptomics, metabolomics, and the microbiome. These have been focused on the response to three common asthma medications: short-acting beta agonists, inhaled corticosteroids, and leukotriene receptor antagonists. Novel associations of different biomarkers with asthma treatment response have been described. However, stronger evidence and more consistent results are required to implement these molecular biomarkers into clinical practice by establishing the most appropriate therapy for each patient.
- asthma
- genomics
- transcriptomics
- epigenomics
- metabolomics
- microbiome
- omics
- inhaled corticosteroid
- short-acting beta agonist
- leukotriene receptor antagonist
Introduction
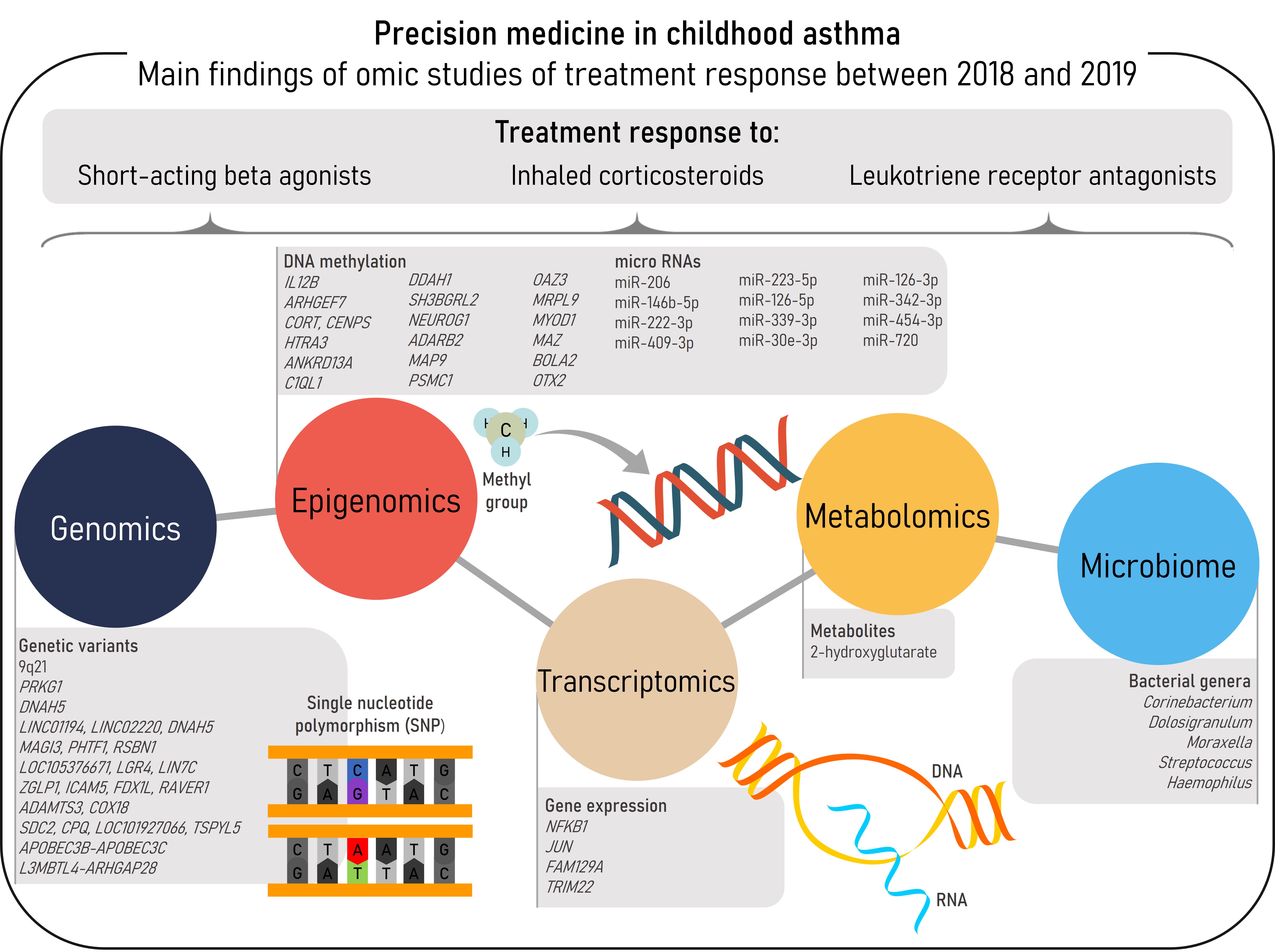
Asthma, the most prevalent chronic respiratory disease in children, is a heterogeneous and complex disease characterized by airway inflammation, reversible airflow obstruction, and bronchial hyperreactivity [1][2]. The most characteristic asthma symptoms are wheeze, cough, chest tightness, and shortness of breath. These symptoms may be prevented, relieved, and controlled by common asthma medications, such as short-acting beta agonists (SABAs), inhaled corticosteroids (ICSs), or leukotriene receptor antagonists (LTRAs) [2]. However, some patients do not achieve adequate control of the disease even with the most intense therapies, such as ICSs at high-doses or oral corticosteroids (OCSs) [3]. These patients are more prone to develop asthma exacerbations, worse episodes of the disease which carry a burden to the patients, their families, and the community [2][4]. Different phenotypes and endotypes of asthma have been described, each one with both shared and differentiated features including the response to pharmacological therapies. Nonetheless, phenotype-guided treatments are scarce and a clear relationship between asthma phenotypes and treatment response has not been established [2]. In this context omic sciences, which consist of high-throughput analyses at different biological layers, would aid to disentangle the molecular mechanisms underlying asthma and treatment response to move toward precision medicine [5]. This entry aims to summarize the findings of omic studies of asthma treatment response conducted between 2018-2019 in pediatric populations (Figure 1).
Summary of the main findings of omics studies of treatment response in childhood asthma within the reviewed period.
Pharmacogenomics
Pharmacogenomics investigates the association of genetic markers across the genome with treatment response. This approach usually focuses on single nucleotide polymorphisms (SNPs), which are changes in the DNA sequence affecting one nucleotide shared by >1% of the population. This type of marker is usually coinherited with other genetic variants due to the linkage disequilibrium (LD) patterns allowing the indirect study of millions of genetic variants that are inferred from hundreds of thousands of SNPs assessed by genotyping arrays. Thus, common genome-wide genetic variation can be studied without any prior hypothesis by genome-wide association studies (GWAS) [6]. Recently, whole-genome sequencing (WGS) emerged as a high-resolution method to study both common and rare genetic variation. Although WGS detects genetic variation not tackled by genome-wide genotyping arrays, its high cost has still limited its application to pharmacogenomic studies [7].
Recent pharmacogenomic studies of childhood asthma have not been limited to European-descent populations as in the past [8][9], but they have mostly analyzed two minority populations from the United States with a high prevalence of asthma and low treatment response [10]: African Americans and Hispanics/Latinos. Specifically, several publications within the period reviewed have been focused on two studies: Study of African Americans, Asthma, Genes and Environments (SAGE) and the Genes-Environment and Admixture in Latino Americans (GALA II) (Table 1). In these studies, genomic analyses of SABA treatment response were carried out by means of GWAS using genotyping arrays and WGS data [11][12]. Additionally, given that African Americans and Hispanics/Latinos are admixed populations, admixture mapping can be applied in order to identify genomic regions in which local ancestry is associated with pharmacogenetic traits [12]. This complementary approach is possible because of the differences in allele frequency of the SNPs depending on their ancestral background [10]. These studies have been focused on bronchodilator drug response (BDR), a measurement of the change in lung function after SABA administration [11][12], and also on the presence/absence of severe asthma exacerbations despite ICS treatment as a proxy of ICS response [13].
Summary of the genomic regions identified by pharmacogenomic studies in childhood asthma performed during 2018-2019.
| rsID/ Chr. Band |
Chr:Positiona | Gene/ Nearest Gene |
Effect Allele | Effect | p-value | Reference |
|---|---|---|---|---|---|---|
| SABA response | ||||||
| rs73650726 | 9:85152666 | 9q21 | A | β=−3.8 | 7.69×10−9 | [12] |
| rs7903366 | 10:53689774 | PRKG1 | T | β=1.23 | 3.94×10−8 | |
| rs7070958 | 10:53691116 | PRKG1 | A | β=−1.24 | 4.09×10−8 | |
| rs7081864 | 10:53690331 | PRKG1 | A | β=1.23 | 4.94×10−8 | |
| rs17834628 | 5:12978566 | LINC01194 LINC02220 DNAH5 | A | OR=1.67 | 1.18×10−8 | [11] |
| rs35661809 | 5:12968341 | LINC01194 LINC02220 DNAH5 | G | OR=1.59 | 3.33×10−8 | |
| 1p13.2 | 1:114177000-1:114178000 | MAGI3 PHTF1 RSBN1 | NA | NA | 4.40×10−9 | |
| 11p14.1 | 11:27507000-11:27508000 | LOC105376671 LGR4 LIN7C | NA | NA | 6.59×10−9 | |
| 19p13.2 | 19:10424000-19:10425000 | ZGLP1 ICAM5 FDX1L RAVER1 | NA | NA | 3.12×10−11 | |
| 4q13.3 | 4:73478000-4:73479000 | ADAMTS3 COX18 | NA | NA | 6.25×10−8 | |
| 8q22.1 | 8:97926000-8:97927000 | SDC2 CPQ LOC101927066 TSPYL5 | NA | NA | 1.32×10−7 | |
| ICS response | ||||||
| rs5995653 | 22:39404249 | APOBEC3B-APOBEC3C | A | OR=0.70 | 3.31×10−7 | [13] |
| rs62081416 | 18:6605442 | L3MBTL4-ARHGAP28 | A | OR=2.44 | 1.57×10−5 | |
| aPosition based on GRCh37/hg19 build. rsID: reference SNP cluster ID; Chr: chromosome; SABA: short-acting beta agonist; ICS: inhaled corticosteroid; OR: odds ratio; NA: not available. | ||||||
Spear et al. [12] performed a GWAS of BDR in African Americans with asthma from SAGE (n=949) analyzing imputed data from genotyping arrays. A SNP from 9q21 (rs73650726) was genome-wide significantly associated with BDR exclusively in African-admixed populations. Moreover, a trans-ethnic meta-GWAS across African American and Hispanic/Latino children and youth (n = 2779) identified genome-wide associations of three SNPs from the protein kinase cGMP-dependent 1 (
PRKG1) gene with SABA response. Interestingly,
PRKG1 is a biologically plausible gene for SABA response given that it encodes a protein involved in the nitric oxide/cGMP signaling pathway, acts as an important regulator of airway inflammation in response to SABA, and participates in smooth muscle relaxation [14,15]. Furthermore,is a biologically plausible gene for SABA response given that it encodes a protein involved in the nitric oxide/cGMP signaling pathway, acts as an important regulator of airway inflammation in response to SABA, and participates in smooth muscle relaxation [14][15]. Furthermore,
PRKG1 has been associated with lung function measurements and asthma risk [16,17]. However, these results need to be replicated in independent studies.has been associated with lung function measurements and asthma risk [16][17]. However, these results need to be replicated in independent studies.
A WGS study was conducted in the same populations selecting patients with asthma with extreme BDR values among African Americans, Puerto Ricans, and Mexicans (n = 1441) [11]. This revealed two SNPs located near the dynein axonemal heavy chain 5 (
DNAH5)gene as significantly associated with BDR in a trans-ethnic meta-analysis.
DNAH5 is ATPase protein that participates in a complex associated with the microtubules. Variants of this gene have been associated with allergic sensitization, lung function, and immunoglobulin E (IgE) serum levels [18,19,20]. The WGS study by Mak et al. [11] also revealed population-specific associations of both common and rare variants (1p13.2 and 11p14.1 in Mexicans and 19p13.2 in African Americans) (Table 1).is ATPase protein that participates in a complex associated with the microtubules. Variants of this gene have been associated with allergic sensitization, lung function, and immunoglobulin E (IgE) serum levels [18][19][20]. The WGS study by Mak et al. [11] also revealed population-specific associations of both common and rare variants (1p13.2 and 11p14.1 in Mexicans and 19p13.2 in African Americans) (Table 1).
Finally, an additional study on the GALA II and SAGE populations evaluated the ICS response in a subset of Hispanic/Latino and African American asthma patients treated with ICSs [13]. In this study, ICS response was defined as the presence/absence of asthma exacerbations in the past 12 months despite ICS treatment [13] (Table 1). Asthma exacerbations were defined by any of the following events originated from asthma symptoms: emergency room (ER) visits, hospitalizations, or OCS use. The association of a SNP from the intergenic region of the genes encoding for the apolipoprotein B mRNA editing enzyme catalytic subunits 3B and 3C (
APOBEC3B-APOBEC3C) was revealed, a result that was replicated in European children with asthma (n=1697). However, this SNP did not reach the genome-wide significance threshold in a meta-analysis across all populations. Despite this, a consistent association of the SNP from thewas replicated in European children with asthma (n = 1697). However, this SNP did not reach the genome-wide significance threshold in a meta-analysis across all populations. Despite this, a consistent association of the SNP from the
APOBEC3B-APOBEC3Cintergenic regions was found with the change in the forced expiratory volume in the first second (FEV
1) after ICS treatment during 6 weeks in a subset of patients with such data available (n=166), as an alternative measurement of ICS response.) after ICS treatment during 6 weeks in a subset of patients with such data available (n = 166), as an alternative measurement of ICS response.
APOBEC3Band
APOBEC3C aresubunits of a cytidine deaminase that carries out RNA editing and participates in the immune response to viruses. In the same study [13], an attempt of replication of genomic regions previously associated with ICS response was performed. The association of the region that included the
L3MBTL4 and ARHGAP28was validated, which has also been associated with post-bronchodilator lung function [18].
[18].
Epigenomics
Epigenetics is the study of the mechanisms involved in gene expression regulation without modifying the DNA sequence. These include DNA methylation (DNAm), microRNA (miRNA) regulation, and histone modifications. Nowadays, whole-genome epigenetic changes can be analyzed through high-throughput techniques. DNAm is the most studied epigenetic modification, involving the methylation of a cytosine base followed by a guanine (CpG site) [21][22][23].
Two epigenome-wide association studies (EWAS) were performed during the period reviewed evaluating the effects of ICS response on DNA methylation patterns in peripheral blood cells (PBCs) [24][25]. In the first study [24], non-Hispanic whites from the Childhood Asthma Management Program (CAMP, n = 154), Europeans from the Children, Allergy, Milieu, Stockholm, Epidemiology (BAMSE, n = 72), and Hispanic/Latinos from the Genetic Epidemiology of Asthma in Costa Rica Study (GACRS, n = 168) were analyzed. Treatment response was measured as two outcomes related to asthma exacerbations in the past 12 months while the patients were treated with ICSs. Hypomethylation of a CpG site near interleukin 12B (
IL12B)was protective for severe exacerbations in a meta-analysis across non-Hispanic whites and Hispanic/Latinos. and this hypomethylation was correlated with lower
IL12Bexpression in blood cells in one of the studies.
IL12B encodes a subunit of two cytokines (IL-12 and IL-23) involved in immune response and airway hyperresponsiveness, and its expression level has been associated with corticosteroids response in bronchial biopsies from asthma patients [26,27,28]. Moreover, in the same study, Wangencodes a subunit of two cytokines (IL-12 and IL-23) involved in immune response and airway hyperresponsiveness, and its expression level has been associated with corticosteroids response in bronchial biopsies from asthma patients [26][27][28]. Moreover, in the same study, Wang
et al. found 13 CpG sites that were significantly associated with the absence of OCS use in a meta-analysis across non-Hispanic whites (n = 322). An interaction analysis identified that hypermethylation of a CpG near the genes encoding for the cortistatin (
CORT)and centromere protein S (
CENPS)was associated with the absence of OCSs use, and also with higher
CORT expression in CAMP [26,27,28]. Interestingly, cortistatin is a peptid involved in the anti-inflammatory process regulated by the hypothalamic-pituitary-adrenal axis and regulates endogenous corticosteroid levels [29,30].expression in CAMP [26][27][28]. Interestingly, cortistatin is a peptid involved in the anti-inflammatory process regulated by the hypothalamic-pituitary-adrenal axis and regulates endogenous corticosteroid levels [29][30].
Summary of the main findings of epigenomic studies focused on DNAm in childhood asthma performed during 2018-2019.
| CpG | Chromosome:Positiona | Gene/Nearest Gene | β | p-value | Reference |
|---|---|---|---|---|---|
| ICS response | |||||
| cg00066816 | 5:158758353 | IL12B | −3.101 | 0.002 | [24] |
| cg00557354 | 13:111767899 | ARHGEF7 | −3.490 | 0.001 | |
| cg04256470 | 1:10510465 | CORT CENPS | 3.620 | <0.001 | |
| cg09495977 | 4:8271507 | HTRA3 | −2.420 | 0.017 | |
| cg12333095 | 12:110437035 | ANKRD13A | −3.485 | 0.001 | |
| cg13818573 | 17:43045372 | C1QL1 | −3.596 | <0.001 | |
| cg21589280 | 1:85930152 | DDAH1 | −3.063 | 0.003 | |
| cg03080985 | 6:80340683 | SH3BGRL2 | −3.077 | 0.003 | |
| cg04330449 | 5:134871166 | NEUROG1 | −2.646 | 0.009 | |
| cg05307923 | 10:1779667 | ADARB2 | −2.577 | 0.011 | |
| cg08724517 | 4:156298205 | MAP9 | 2.951 | 0.004 | |
| cg11665562 | 14:90723462 | PSMC1 | −3.250 | 0.001 | |
| cg14269514 | 1:151736130 | OAZ3 MRPL9 | −3.112 | 0.002 | |
| cg24322623 | 11:17740431 | MYOD1 | −2.964 | 0.004 | |
| cg27254601 | 16:29817104 | MAZ BOLA2 | 3.598 | 0.0005 | [25] |
| cg15607672 | 14:57277228 | OTX2 | 2.123 | 0.0363 | |
| aPosition based on GRCh37/hg19 build. DNAm: DNA methylation; ICS: inhaled corticosteroid. | |||||
The second EWAS of ICS response was performed in non-Hispanic white children from CAMP (n = 152) analyzing the change in FEV
1after eight weeks of ICS treatment [25]. Relative hypermethylation of a CpG from the bolA family member 2 (
BOLA2) genewas associated with lung function improvement and with increased gene expression of
BOLA2. BOLA2 is involved in the maturation process of iron-sulfur containing proteins [31]. Gene expression levels of
BOLA2 in airway cells differ by asthma status [29], and intronic variants of this gene are associated with eosinophil levels and lung function measurements [17,32,33]. Additionally, the study by Wangin airway cells differ by asthma status [29], and intronic variants of this gene are associated with eosinophil levels and lung function measurements [17][32][33]. Additionally, the study by Wang
etal. validated the hypermethylation of the orthodenticle homeobox 2 (
OTX2) gene in PBCs, which had been previously associated with good response to OCS in nasal cells [34].gene in PBCs, which had been previously associated with good response to OCS in nasal cells [34].
Regarding miRNA profiling without a prior hypothesis, one study has been performed between 2018-2019. Kho
etal. evaluated the association of 754 circulating miRNAs in serum with ICS response in non-Hispanic white children with asthma from CAMP (n = 153) [35]. A total of 12 miRNAs were associated with the risk of exacerbations, defined as steroid bursts in the past year because of asthma ICS treatment (Table 3). The strongest association was described for miR-206, which had higher serum expression levels in non-exacerbators compared to exacerbators. In addition, a predictive model for asthma exacerbations based on three miRNAs (miR-206, miR-146b-5p, and miR-720) and clinical variables had an improved prediction area under the receiver characteristics curve (AUC) than a model exclusively based on clinical parameters (0.81
vs0.67). Strikingly, these three miRNAs have been related to asthma in cellular and animal models [35], and miR-146b-5b and miR-206 had also been associated with baseline FEV
1/FVC [36]. Moreover, four biological pathways regulated by the three miRNAs were revealed by Kho
et al., including two previously associated with asthma [35].al., including two previously associated with asthma [35].
Summary of findings of the only epigenomic study focused on the association of circulating miRNA with inhaled corticosteroid (ICS) response in childhood asthma during 2018-2019 [35].
| miRNA | Odds Ratio | p-value |
|---|---|---|
| miR-206 | 0.60 | 0.004 |
| miR-146b-5p | 0.66 | 0.007 |
| miR-222-3p | 0.70 | 0.02 |
| miR-409-3p | 0.73 | 0.02 |
| miR-223-5p | 0.62 | 0.02 |
| miR-126-5p | 0.68 | 0.03 |
| miR-339-3p | 0.72 | 0.03 |
| miR-30e-3p | 0.70 | 0.03 |
| miR-126-3p | 0.74 | 0.03 |
| miR-342-3p | 0.80 | 0.04 |
| miR-454-3p | 0.77 | 0.04 |
| miR-720 | 0.71 | 0.046 |
| miRNA: micro ribonucleic acid. | ||
Transcriptomics
Transcriptomics is the study of all RNA transcripts expressed in a cell or individual by high-throughput methods such as RNA sequencing (RNA-seq) or microarrays. During 2018-2019, most transcriptomic studies of treatment response in pediatric asthma focused on ICS response. A microarray analysis of peripheral blood mononuclear cells (PBMCs) from Taiwanese children with asthma (n = 133) showed that patients with poor asthma control have a specific transcriptomic pattern associated with glucocorticoid signaling and immune response [37]. Moreover, the application of system biology approaches in the analysis of transcriptomic data of ICS response in non-Hispanic white children from CAMP has been performed by two studies. In the first study, Qui et al. [38] analyzed gene expression networks in immortalized B-cell lines from children treated with ICSs during two months and classified as good (n = 47) or poor (n = 48) ICS responders based on changes in lung function after ICS treatment. While good responders had an enrichment in immune response and proapoptotic corticosteroid-induced pathways, poor responders showed enrichment in antiapoptotic pathways. Additionally, two transcription factors (TFs), the nuclear factor kappa B subunit 1 (
NFKB1)and the Jun proto-oncogene, AP-1 transcription factor subunit (
JUN),had differential regulation between both groups. Interestingly, dysregulation of
NFKB1is associated with inflammatory diseases and inadequate immune cell development, and
JUN has been related to macrophage activation [39].has been related to macrophage activation [39].
McGeachie et al. [40] conducted a multi-omic analysis in non-Hispanic white children with asthma from CAMP treated with budesonide (n = 104). Treatment response was evaluated by a composite phenotype of ICS response, which was integrated with genome-wide genotyping data and the response of immortalized lymphoblastoid cells to dexamethasone into a network. A total of seven genes were associated with steroid response and four of them were selected for in vitro functional analyses in lung epithelial cells. A knockdown of the family with sequence similarity 129 member A (
FAM129A)gene showed a
reduced response to dexamethasone.
FAM129A encodes for a protein involved in an apoptosis pathway [41].encodes for a protein involved in an apoptosis pathway [41].
Katayama et al. [42] performed a transcriptomic study of LTRA response in children aged 6–48 months recruited during an acute wheezing episode and followed-up to seven years (n = 107). A weighted gene co-expression network analysis (WGCNA) [43] was performed in leukocytes to identify modules of genes involved in LTRA response. A module of 145 co-regulated genes involved in interferon signaling pathways, inflammation, and antiviral response was correlated with acute wheezing. Moreover, this module was associated with lung function and the number of asthma exacerbations, and had a good predictive value for future LTRA medication (AUC = 0.81). The tripartite motif containing 22 (
TRIM22)gene showed the strongest association with LTRA treatment. Interestingly,
TRIM22 is involved in the antiviral response regulated by one of the interferon pathways [44].Metabolomics
Metabolomics provides the profile of the whole metabolite composition (metabolome) in biological samples by means of high-throughput analytical techniques, such as mass spectrophotometry, nuclear magnetic resonance, or spectroscopic methods. interestingly, metabolomics can be performed in non-invasive samples such as exhaled air (breathomics) and this recent omic has been applied to asthma with promising results [45][46][47].
Kelly et al. examined the interaction of age and 501 serum metabolites on BDR after albuterol administration in blood samples from children with asthma from CAMP at three-time points every four years, with mean ages of 8.8 (n = 560), 12.8 (n = 563), and 16.8 (n = 295), respectively [48]. A total of 39 metabolites, mainly lipids, showed a nominal interaction with age on BDR, being the strongest interaction observed for the 2-hydroxyglutarate. Results of this metabolite were replicated in Hispanic children with asthma from GACRS (n = 320), with a mean age of 9.1 years. However, these results would not be considered significant after multiple comparison adjustment.
Kelly et al. also conducted a multi-omic study of lung function in Hispanic/Latino children with asthma (n = 325) simultaneously analyzing gene transcripts and metabolites with WGCNA [49]. Four transcript modules and five metabolite clusters were found to be related to lung function and seven of them were found to interact among them. Interestingly, one transcriptomic module was associated with BDR and with a lipid metabolomic module, and had as a hub the well-known pediatric-asthma loci
ORMDL3, which is involved in sphingolipid biosynthesis [50,51,52]. Furthermore, a SNP frominvolved in sphingolipid biosynthesis [50][51][52]. Furthermore, a SNP from
ORMDL3was found to be an eQTL of
ORMDL3 and was associated with several lipids included in the metabolomic module. Reinforcing these results, findings were replicated in children with asthma from CAMP (n=207).and was associated with several lipids included in the metabolomic module. Reinforcing these results, findings were replicated in children with asthma from CAMP (n = 207).
Microbiome
The study of the microbiome involves the analysis of the genetic composition of the microbial communities, known as microbiota. The advances of next-generation sequencing (NGS) techniques have allowed the extension of this type of study by means of targeted sequencing techniques, such as bacterial profiling analyzing the 16S ribosomal RNA gene (16S rRNA), or metagenomic approaches [53][54].
Zhou et al. [55] performed a longitudinal study to identify changes in nasal microbiota related to asthma exacerbations risk despite ICS treatment in European children with mild–moderate persistent asthma (n = 214) as part of a clinical trial. The nasal bacterial microbiome was characterized by means of 16S rRNA sequencing by collecting nasal swabs at the time of well-controlled asthma (randomization) and during the first episode of loss of asthma control. A nasal microbiome dominated by
Corynebacteriumand
Dolosigranulumat randomization was associated with fewer episodes of early loss of asthma control and longer times to develop such episodes. Additionally, during the first loss-of-asthma-control episode,
Streptococcuswas the most prevalent dominant genus in the nasal microbiome. Moreover, higher bacterial richness and total bacterial load were observed during asthma control loss episode. Furthermore,
Corynebacteriumwas protective for exacerbations requiring OCS use, and the predominance of
Moraxella instead of Corynebacterium + Dolosigranulum,was associated with the highest risk of exacerbations. Moreover,
Moraxella,
Streptococcus, and
Haemophilus are pathogens commonly found in the nasal microbiome of asthma patients [56,57,58], andare pathogens commonly found in the nasal microbiome of asthma patients [56][57][58], and
Moraxellahas also been associated with a higher risk of asthma exacerbations [59]. In contrast,
Corynebacterium is the most abundant commensal bacteria in the nasal microbiome from healthy individuals, and asthma patients show decreased relative abundance of this bacteria [56,60].is the most abundant commensal bacteria in the nasal microbiome from healthy individuals, and asthma patients show decreased relative abundance of this bacteria [56][60].
Conclusions
References
Omic studies have allowed the identification of novel biomarkers associated with the response to asthma medications in children. However, some findings have not been validated across populations and fail to fully explain the differences in treatment response in childhood asthma. This is expected since asthma is a complex and multifactorial disease, and different phenotypes may not share the same underlying mechanisms. However, further advances in omic studies will aid to understand how different biological layers are implicated in disease pathogenesis and also how they could interact among them to determine the response to pharmacological therapies.
- The Global Asthma Report 2018; Global Asthma Network: Auckland, New Zealand, 2018; ISBN 978-0-473-29125-9\r978-0-473-29126-6.
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention; Global Initiative for Asthma: Fontana, WI, USA, 2019. [Google Scholar]
- Guilbert, T.W.; Bacharier, L.B.; Fitzpatrick, A.M. Severe Asthma in Children. J. Allergy Clin. Immunol. Pract. 2014, 2, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Pereira, A.M.; Morais-Almeida, M. Asthma Costs and Social Impact. Asthma Res. Pract. 2017, 3, 1. [Google Scholar] [CrossRef]
- Tyler, S.R.; Bunyavanich, S. Leveraging -Omics for Asthma Endotyping. J. Allergy Clin. Immunol. 2019, 144, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef]
- Petersen, B.S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A. Opportunities and Challenges of Whole-Genome and -Exome Sequencing. BMC Genet. 2017, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Luis, E.; Hernandez-Pacheco, N.; Vijverberg, S.J.; Flores, C.; Pino-Yanes, M. Role of Genomics in Asthma Exacerbations. Curr. Opin. Pulm. Med. 2019, 25, 101–112. [Google Scholar] [CrossRef]
- García-Menaya, J.M.; Cordobés-Durán, C.; García-Martín, E.; Agúndez, J.A.G. Pharmacogenetic Factors Affecting Asthma Treatment Response. Potential Implications for Drug Therapy. Front. Pharmacol. 2019, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pacheco, N.; Flores, C.; Oh, S.S.; Burchard, E.G.; Pino-Yanes, M. What Ancestry Can Tell Us About the Genetic Origins of Inter-Ethnic Differences in Asthma Expression. Curr. Allergy Asthma Rep. 2016, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.C.Y.; White, M.J.; Eckalbar, W.L.; Szpiech, Z.A.; Oh, S.S.; Pino-Yanes, M.; Hu, D.; Goddard, P.; Huntsman, S.; Galanter, J.; et al. Whole-Genome Sequencing of Pharmacogenetic Drug Response in Racially Diverse Children with Asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.L.; Hu, D.; Pino-Yanes, M.; Huntsman, S.; Eng, C.; Levin, A.M.; Ortega, V.E.; White, M.J.; McGarry, M.E.; Thakur, N.; et al. A Genome-Wide Association and Admixture Mapping Study of Bronchodilator Drug Response in African Americans with Asthma. Pharmacogenomics J. 2019, 19, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pacheco, N.; Farzan, N.; Francis, B.; Karimi, L.; Repnik, K.; Vijverberg, S.J.; Soares, P.; Schieck, M.; Gorenjak, M.; Forno, E.; et al. Genome-Wide Association Study of Inhaled Corticosteroid Response in Admixed Children with Asthma. Clin. Exp. Allergy 2019, 49, 789–798. [Google Scholar] [CrossRef]
- Kanazawa, H.; Hirata, K.; Yoshikawa, J. Nitrogen Oxides Reduce Albuterol-Induced Bronchodilation in Patients with Bronchial Asthma. Respiration 2002, 69, 490–495. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.M. Multiple Roles of Nitric Oxide in the Airways. Thorax 2003, 58, 175–182. [Google Scholar] [CrossRef]
- Ferreira, M.A.R.; Matheson, M.C.; Duffy, D.L.; Marks, G.B.; Hui, J.; Le Souëf, P.; Danoy, P.; Baltic, S.; Nyholt, D.R.; Jenkins, M.; et al. Identification of IL6R and Chromosome 11q13.5 as Risk Loci for Asthma. Lancet 2011, 378, 1006–1014. [Google Scholar] [CrossRef]
- Kichaev, G.; Bhatia, G.; Loh, P.R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef]
- Lutz, S.M.; Cho, M.H.; Young, K.; Hersh, C.P.; Castaldi, P.J.; McDonald, M.L.; Regan, E.; Mattheisen, M.; DeMeo, D.L.; Parker, M.; et al. A Genome-Wide Association Study Identifies Risk Loci for Spirometric Measures among Smokers of European and African Ancestry. BMC Genet. 2015, 16, 138. [Google Scholar] [CrossRef]
- Lee, J.H.; McDonald, M.L.; Cho, M.H.; Wan, E.S.; Castaldi, P.J.; Hunninghake, G.M.; Marchetti, N.; Lynch, D.A.; Crapo, J.D.; Lomas, D.A.; et al. DNAH5 Is Associated with Total Lung Capacity in Chronic Obstructive Pulmonary Disease. Respir. Res. 2014, 15, 97. [Google Scholar] [CrossRef]
- Ramasamy, A.; Curjuric, I.; Coin, L.J.; Kumar, A.; McArdle, W.L.; Imboden, M.; Leynaert, B.; Kogevinas, M.; Schmid-Grendelmeier, P.; Pekkanen, J.; et al. A Genome-Wide Meta-Analysis of Genetic Variants Associated with Allergic Rhinitis and Grass Sensitization and Their Interaction with Birth Order. J. Allergy Clin. Immunol. 2011, 128, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Forno, E.; Celedón, J.C. Epigenomics and Transcriptomics in the Prediction and Diagnosis of Childhood Asthma: Are We There Yet? Front. Pediatr. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.J.; Greally, J.M. Introduction to Epigenomics and Epigenome-Wide Analysis. Methods Mol. Biol. 2010, 620, 243–265. [Google Scholar] [CrossRef]
- Kronfol, M.M.; Dozmorov, M.G.; Huang, R.; Slattum, P.W.; McClay, J.L. The Role of Epigenomics in Personalized Medicine. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 33–45. [Google Scholar] [CrossRef]
- Wang, A.L.; Gruzieva, O.; Qiu, W.; Kebede Merid, S.; Celedón, J.C.; Raby, B.A.; Söderhäll, C.; DeMeo, D.L.; Weiss, S.T.; Melén, E.; et al. DNA Methylation Is Associated with Inhaled Corticosteroid Response in Persistent Childhood Asthmatics. Clin. Exp. Allergy 2019, 49, 1225–1234. [Google Scholar] [CrossRef]
- Wang, A.L.; Qiu, W.; Demeo, D.L.; Raby, B.A.; Weiss, S.T.; Tantisira, K.G. DNA Methylation Is Associated with Improvement in Lung Function on Inhaled Corticosteroids in Pediatric Asthmatics. Pharmacogenet. Genom. 2019, 29, 65–68. [Google Scholar] [CrossRef]
- Szabo, S.J.; Jacobson, N.G.; Dighe, A.S.; Gubler, U.; Murphy, K.M. Developmental Commitment to the Th2 Lineage by Extinction of IL-12 Signaling. Immunity 1995, 2, 665–675. [Google Scholar] [CrossRef]
- Naseer, T.; Minshall, E.M.; Leung, D.Y.M.; Laberge, S.; Ernst, P.; Martin, R.J.; Hamid, Q. Expression of IL-12 and IL-13 MRNA in Asthma and Their Modulation in Response to Steroid Therapy. Am. J. Respir. Crit. Care Med. 1997, 155, 845–851. [Google Scholar] [CrossRef]
- Keane-Myers, A.; Wysocka, M.; Trinchieri, G.; Wills-Karp, M. Resistance to Antigen-Induced Airway Hyperresponsiveness Requires Endogenous Production of IL-12. J. Immunol. 1998, 161, 919–926. [Google Scholar]
- Giordano, R.; Picu, A.; Bonelli, L.; Broglio, F.; Prodam, F.; Grottoli, S.; Muccioli, G.; Ghigo, E.; Arvat, E. The Activation of Somatostatinergic Receptors by Either Somatostatin-14 or Cortistatin-17 Often Inhibits ACTH Hypersecretion in Patients with Cushing’s Disease. Eur. J. Endocrinol. 2007, 157, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, E.; Pedreño, M.; Delgado-Maroto, V.; Souza-Moreira, L.; Delgado, M. Lulling Immunity, Pain, and Stress to Sleep with Cortistatin. Ann. N. Y. Acad. Sci. 2015, 1351, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.G.; Palenchar, D.J.; Wildemann, J.D.; Philpott, C.C. A Glutaredoxin-BolA Complex Serves as an Iron-Sulfur Cluster Chaperone for the Cytosolic Cluster Assembly Machinery. J. Biol. Chem. 2016, 291, 22344–22356. [Google Scholar] [CrossRef] [PubMed]
- Bigler, J.; Boedigheimer, M.; Schofield, J.P.R.; Skipp, P.J.; Corfield, J.; Rowe, A.; Sousa, A.R.; Timour, M.; Twehues, L.; Hu, X.; et al. A Severe Asthma Disease Signature from Gene Expression Profiling of Peripheral Blood from U-BIOPRED Cohorts. Am. J. Respir. Crit. Care Med. 2017, 195, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415–1429.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Biagini Myers, J.M.; Yadagiri, V.K.; Ulm, A.; Chen, X.; Weirauch, M.T.; Khurana Hershey, G.K.; Ji, H. Nasal DNA Methylation Differentiates Corticosteroid Treatment Response in Pediatric Asthma: A Pilot Study. PLoS ONE 2017, 12, e0186150. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.T.; McGeachie, M.J.; Moore, K.G.; Sylvia, J.M.; Weiss, S.T.; Tantisira, K.G. Circulating MicroRNAs and Prediction of Asthma Exacerbation in Childhood Asthma. Respir. Res. 2018, 19, 128. [Google Scholar] [CrossRef]
- Kho, A.T.; Sharma, S.; Davis, J.S.; Spina, J.; Howard, D.; McEnroy, K.; Moore, K.; Sylvia, J.; Qiu, W.; Weiss, S.T.; et al. Circulating MicroRNAs: Association with Lung Function in Asthma. PLoS ONE 2016, 11, e0157998. [Google Scholar] [CrossRef]
- Yeh, Y.L.; Su, M.W.; Chiang, B.L.; Yang, Y.H.; Tsai, C.H.; Lee, Y.L. Genetic Profiles of Transcriptomic Clusters of Childhood Asthma Determine Specific Severe Subtype. Clin. Exp. Allergy 2018, 48, 1164–1172. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, F.; Glass, K.; Yuan, G.C.; Quackenbush, J.; Zhou, X.; Tantisira, K.G. Differential Connectivity of Gene Regulatory Networks Distinguishes Corticosteroid Response in Asthma. J. Allergy Clin. Immunol. 2018, 141, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Beste, M.T.; Pfäffle-Doyle, N.; Prentice, E.A.; Morris, S.N.; Lauffenburger, D.A.; Isaacson, K.B.; Griffith, L.G. Endometriosis: Molecular Network Analysis of Endometriosis Reveals a Role for c-Jun-Regulated Macrophage Activation. Sci. Transl. Med. 2014, 6, 222ra16. [Google Scholar] [CrossRef] [PubMed]
- McGeachie, M.J.; Clemmer, G.L.; Hayete, B.; Xing, H.; Runge, K.; Wu, A.C.; Jiang, X.; Lu, Q.; Church, B.; Khalil, I.; et al. Systems Biology and in Vitro Validation Identifies Family with Sequence Similarity 129 Member A (FAM129A) as an Asthma Steroid Response Modulator. J. Allergy Clin. Immunol. 2018, 142, 1479–1488.e12. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, J.; Liu, J.; Huang, Y.; Zhou, Y.; Yang, S.; Zhang, W.; Yang, M.; Zhang, H. Niban Protein Regulates Apoptosis in HK-2 Cells via Caspase-Dependent Pathway. Ren. Fail. 2019, 41, 455–466. [Google Scholar] [CrossRef]
- Katayama, S.; Hammar, K.S.; Krjutškov, K.; Einarsdottir, E.; Hedlin, G.; Kere, J.; Söderhäll, C. Acute Wheeze-Specific Gene Module Shows Correlation with Vitamin D and Asthma Medication. Eur. Respir. J. 2019, 55, 1901330. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Lian, Q.; Sun, B. Interferons Command Trim22 to Fight against Viruses. Cell. Mol. Immunol. 2017, 14, 794–796. [Google Scholar] [CrossRef]
- Pité, H.; Morais-Almeida, M.; Rocha, S.M. Metabolomics in Asthma: Where Do We Stand? Curr. Opin. Pulm. Med. 2018, 24, 94–103. [Google Scholar] [CrossRef]
- Neerincx, A.H.; Vijverberg, S.J.H.; Bos, L.D.J.; Brinkman, P.; van der Schee, M.P.; de Vries, R.; Sterk, P.J.; Maitland-van der Zee, A.-H. Breathomics from Exhaled Volatile Organic Compounds in Pediatric Asthma. Pediatr. Pulmonol. 2017, 52, 1616–1627. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, S.H.; Kim, J.H.; Hwang, S.; Yoo, H.J. Understanding Metabolomics in Biomedical Research. Endocrinol. Metab. 2016, 31, 7–16. [Google Scholar] [CrossRef]
- Kelly, R.S.; Sordillo, J.E.; Lutz, S.M.; Avila, L.; Soto-Quiros, M.; Celedón, J.C.; McGeachie, M.J.; Dahlin, A.; Tantisira, K.; Huang, M.; et al. Pharmacometabolomics of Bronchodilator Response in Asthma and the Role of Age-Metabolite Interactions. Metabolites 2019, 9, 179. [Google Scholar] [CrossRef]
- Kelly, R.S.; Chawes, B.L.; Blighe, K.; Virkud, Y.V.; Croteau-Chonka, D.C.; McGeachie, M.J.; Clish, C.B.; Bullock, K.; Celedón, J.C.; Weiss, S.T.; et al. An Integrative Transcriptomic and Metabolomic Study of Lung Function in Children With Asthma. Chest 2018, 154, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Paulenda, T.; Draber, P. The Role of ORMDL Proteins, Guardians of Cellular Sphingolipids, in Asthma. Allergy Eur. J. Allergy Clin. Immunol. 2016, 71, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, A.A.; Potaczek, D.P.; Schedel, M.; Gersting, S.W.; Michel, S.; Krajnov, N.; Gaertner, V.D.; Klingbeil, J.M.; Illig, T.; Franke, A.; et al. Childhood Asthma Is Associated with Mutations and Gene Expression Differences of ORMDL Genes That Can Interact. Allergy Eur. J. Allergy Clin. Immunol. 2015, 70, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Ono, J.G.; Worgall, T.S.; Worgall, S. 17q21 Locus and ORMDL3: An Increased Risk for Childhood Asthma. Pediatr. Res. 2014, 75, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, G.M. Genomic Approaches to Studying the Human Microbiota. Nature 2012, 489, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the Microbiome: Advantages of Whole Genome Shotgun versus 16S Amplicon Sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jackson, D.; Bacharier, L.B.; Mauger, D.; Boushey, H.; Castro, M.; Durack, J.; Huang, Y.; Lemanske, R.F.; Storch, G.A.; et al. The Upper-Airway Microbiota and Loss of Asthma Control among Asthmatic Children. Nat. Commun. 2019, 10, 5714. [Google Scholar] [CrossRef]
- Durack, J.; Huang, Y.J.; Nariya, S.; Christian, L.S.; Mark Ansel, K.; Beigelman, A.; Castro, M.; Dyer, A.M.; Israel, E.; Kraft, M.; et al. Bacterial Biogeography of Adult Airways in Atopic Asthma. Microbiome 2018, 6, 104. [Google Scholar] [CrossRef]
- Chung, K.F. Airway Microbial Dysbiosis in Asthmatic Patients: A Target for Prevention and Treatment? J. Allergy Clin. Immunol. 2017, 139, 1071–1081. [Google Scholar] [CrossRef]
- Depner, M.; Ege, M.J.; Cox, M.J.; Dwyer, S.; Walker, A.W.; Birzele, L.T.; Genuneit, J.; Horak, E.; Braun-Fahrländer, C.; Danielewicz, H.; et al. Bacterial Microbiota of the Upper Respiratory Tract and Childhood Asthma. J. Allergy Clin. Immunol. 2017, 139, 826–834.e13. [Google Scholar] [CrossRef]
- McCauley, K.; Durack, J.; Valladares, R.; Fadrosh, D.W.; Lin, D.L.; Calatroni, A.; LeBeau, P.K.; Tran, H.T.; Fujimura, K.E.; LaMere, B.; et al. Distinct Nasal Airway Bacterial Microbiotas Differentially Relate to Exacerbation in Pediatric Patients with Asthma. J. Allergy Clin. Immunol. 2019, 144, 1187–1197. [Google Scholar] [CrossRef]
- Zhou, Y.; Gao, H.; Mihindukulasuriya, K.A.; Rosa, P.S.L.; Wylie, K.M.; Vishnivetskaya, T.; Podar, M.; Warner, B.; Tarr, P.I.; Nelson, D.E.; et al. Biogeography of the Ecosystems of the Healthy Human Body. Genome Biol. 2013, 14, R1. [Google Scholar] [CrossRef]
References
- The Global Asthma Report 2018; Global Asthma Network: Auckland, New Zealand, 2018; ISBN 978-0-473-29125-9\r978-0-473-29126-6.
- Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention; Global Initiative for Asthma: Fontana, WI, USA, 2019.
- Guilbert, T.W.; Bacharier, L.B.; Fitzpatrick, A.M; Severe Asthma in Children. J. Allergy Clin. Immunol. Pract. 2014, 2, 489–500.
- Carlos Nunes; Ana Margarida Pereira; Mário Morais-Almeida; Asthma costs and social impact. Asthma Research and Practice 2017, 3, 1, 10.1186/s40733-016-0029-3.
- Tyler, S.R.; Bunyavanich, S; Leveraging-Omics for Asthma Endotyping. J. Allergy Clin. Immunol. 2019, 144, 13–23.
- Peter M. Visscher; Naomi R. Wray; Qian Zhang; Pamela Sklar; Mark I. McCarthy; Matthew A. Brown; Jian Yang; 10 Years of GWAS Discovery: Biology, Function, and Translation. The American Journal of Human Genetics 2017, 101, 5-22, 10.1016/j.ajhg.2017.06.005.
- Petersen, B.S.; Fredrich, B.; Hoeppner, M.P.; Ellinghaus, D.; Franke, A; Opportunities and Challenges of Whole-Genome and-Exome Sequencing. BMC Genet 2017, 18, 14.
- Esther Herrera-Luis; Natalia Hernandez-Pacheco; Susanne J. Vijverberg; Carlos Flores; Maria Pino-Yanes; Role of genomics in asthma exacerbations. Current Opinion in Pulmonary Medicine 2019, 25, 101-112, 10.1097/mcp.0000000000000533.
- García-Menaya, J.M.; Cordobés-Durán, C.; García-Martín, E.; Agúndez, J.A.G; Pharmacogenetic Factors Affecting Asthma Treatment Response. Potential Implications for Drug Therapy. Front. Pharmacol. 2019, 10, 520.
- Natalia Hernandez-Pacheco; Carlos Flores; Sam S. Oh; Esteban G. Burchard; Maria Pino-Yanes; What Ancestry Can Tell Us About the Genetic Origins of Inter-Ethnic Differences in Asthma Expression. Current Allergy and Asthma Reports 2016, 16, 53, 10.1007/s11882-016-0635-4.
- Mak, A.C.Y.; White, M.J.; Eckalbar, W.L.; Szpiech, Z.A.; Oh, S.S.; Pino-Yanes, M.; Hu, D.; Goddard, P.; Huntsman, S.; Galanter, J.; et al.et al Faculty Opinions recommendation of Whole-Genome Sequencing of Pharmacogenetic Drug Response in Racially Diverse Children with Asthma.. Am. J. Respir. Crit. Care Med. 2018, 197, 1552–1564, 10.3410/f.732806514.793547058.
- Spear, M.L.; Hu, D.; Pino-Yanes, M.; Huntsman, S.; Eng, C.; Levin, A.M.; Ortega, V.E.; White, M.J.; McGarry, M.E.; Thakur, N.; et al.et al A Genome-Wide Association and Admixture Mapping Study of Bronchodilator Drug Response in African Americans with Asthma. Pharmacogenomics J. 2019, 19, 249–259.
- Hernandez-Pacheco, N.; Farzan, N.; Francis, B.; Karimi, L.; Repnik, K.; Vijverberg, S.J.; Soares, P.; Schieck, M.; Gorenjak, M.; Forno, E.; et al.et al Genome-wide association study of inhaled corticosteroid response in African-admixed children with asthma. Clin. Exp. Allergy 2019, 49, 789–798.
- Hiroshi Kanazawa; Kazuto Hirata; Junichi Yoshikawa; Nitrogen oxides reduce albuterol-induced bronchodilation in patients with bronchial asthma.. Respiration 2002, 69, 490-495, 10.1159/000066466.
- Fabio L.M. Ricciardolo; Multiple roles of nitric oxide in the airways. Thorax 2003, 58, 175–182.
- Ferreira, M.A.R.; Matheson, M.C.; Duffy, D.L.; Marks, G.B.; Hui, J.; Le Souëf, P.; Danoy, P.; Baltic, S.; Nyholt, D.R.; Jenkins, M.; et al.et al Identification of IL6R and Chromosome 11q13.5 as Risk Loci for Asthma. Lancet 2011, 378, 1006–1014.
- Kichaev, G.; Bhatia, G.; Loh, P.R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L; Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75.
- Lutz, S.M.; Cho, M.H.; Young, K.; Hersh, C.P.; Castaldi, P.J.; McDonald, M.L.; Regan, E.; Mattheisen, M.; DeMeo, D.L.; Parker, M.; et al.et al A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry.. BMC Genetics 2015, 16, 138, 10.1186/s12863-015-0299-4.
- Lee, J.H.; McDonald, M.L.; Cho, M.H.; Wan, E.S.; Castaldi, P.J.; Hunninghake, G.M.; Marchetti, N.; Lynch, D.A.; Crapo, J.D.; Lomas, D.A.; et al.et al DNAH5 is associated with total lung capacity in chronic obstructive pulmonary disease.. Respiratory Research 2014, 15, 97, 10.1186/s12931-014-0097-y.
- Ramasamy, A.; Curjuric, I.; Coin, L.J.; Kumar, A.; McArdle, W.L.; Imboden, M.; Leynaert, B.; Kogevinas, M.; Schmid-Grendelmeier, P.; Pekkanen, J.; et al.et al A genome-wide meta-analysis of genetic variants associated with allergic rhinitis and grass sensitization and their interaction with birth order. Journal of Allergy and Clinical Immunology 2011, 128, 996-1005, 10.1016/j.jaci.2011.08.030.
- Forno, E.; Celedón, J.C. Epigenomics and Transcriptomics in the Prediction and Diagnosis of Childhood Asthma: Are We There Yet? Front. Pediatr. 2019, 7, 115.
- Melissa Fazzari; John M. Greally; Introduction to Epigenomics and Epigenome-Wide Analysis. Advanced Structural Safety Studies 2010, 620, 243-265, 10.1007/978-1-60761-580-4_7.
- Mohamad M. Kronfol; Mikhail G. Dozmorov; Rong Huang; Patricia W. Slattum; Joseph L. McClay; The role of epigenomics in personalized medicine. Expert Review of Precision Medicine and Drug Development 2017, 2, 33-45, 10.1080/23808993.2017.1284557.
- Alberta Wang; Olena Gruzieva; Weiliang Qiu; Simon Kebede Merid; Juan C. Celedón; Benjamin A. Raby; Cilla Söderhäll; Dawn L. DeMeo; Scott T. Weiss; Erik Melén; et al.Kelan G Tantisira DNA methylation is associated with inhaled corticosteroid response in persistent childhood asthmatics. Clinical Experimental Allergy 2019, 49, 1225-1234, 10.1111/cea.13447.
- Alberta L. Wang; Weiliang Qiu; Dawn L. DeMeo; Benjamin A. Raby; Scott Weiss; Kelan G. Tantisira; DNA methylation is associated with improvement in lung function on inhaled corticosteroids in pediatric asthmatics. Pharmacogenetics and Genomics 2019, 29, 65-68, 10.1097/fpc.0000000000000366.
- Susanne J. Szabo; Nile G. Jacobson; Anand S. Dighe; Uell Gubler; Kenneth M. Murphy; Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity 1995, 2, 665-675, 10.1016/1074-7613(95)90011-x.
- T Naseer; E M Minshall; D Y Leung; S Laberge; P Ernst; R J Martin; Q Hamid; Expression of IL-12 and IL-13 mRNA in asthma and their modulation in response to steroid therapy. American Journal of Respiratory and Critical Care Medicine 1997, 155, 845-851, 10.1164/ajrccm.155.3.9117015.
- A Keane-Myers; M Wysocka; G Trinchieri; M Wills-Karp; Resistance to antigen-induced airway hyperresponsiveness requires endogenous production of IL-12. The Journal of Immunology 1998, 161, 919–926.
- R Giordano; A Picu; L Bonelli; F Broglio; F Prodam; S Grottoli; Giampiero Muccioli; E. Ghigo; Emanuela Arvat; The activation of somatostatinergic receptors by either somatostatin-14 or cortistatin-17 often inhibits ACTH hypersecretion in patients with Cushing’s disease. European Journal of Endocrinology 2007, 157, 393-398, 10.1530/eje-07-0147.
- Elena Gonzalez-Rey; Marta Pedreño; Virginia Delgado-Maroto; Luciana Souza-Moreira; M. Delgado; Lulling immunity, pain, and stress to sleep with cortistatin. Annals of the New York Academy of Sciences 2015, 1351, 89-98, 10.1111/nyas.12789.
- Avery G. Frey; Daniel J. Palenchar; Justin D. Wildemann; Caroline C. Philpott; A Glutaredoxin·BolA Complex Serves as an Iron-Sulfur Cluster Chaperone for the Cytosolic Cluster Assembly Machinery. Journal of Biological Chemistry 2016, 291, 22344-22356, 10.1074/jbc.m116.744946.
- Bigler, J.; Boedigheimer, M.; Schofield, J.P.R.; Skipp, P.J.; Corfield, J.; Rowe, A.; Sousa, A.R.; Timour, M.; Twehues, L.; Hu, X.; et al.et al A Severe Asthma Disease Signature from Gene Expression Profiling of Peripheral Blood from U-BIOPRED Cohorts. American Journal of Respiratory and Critical Care Medicine 2017, 195, 1311-1320, 10.1164/rccm.201604-0866OC.
- Astle, W.J.; Elding, H.; Jiang, T.; Allen, D.; Ruklisa, D.; Mann, A.L.; Mead, D.; Bouman, H.; Riveros-Mckay, F.; Kostadima, M.A.; et al.et al The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 2016, 167, 1415-1429.e19, 10.1016/j.cell.2016.10.042.
- Xue Zhang; Jocelyn M. Biagini Myers; Veda K. Yadagiri; Ashley Ulm; Xiaoting Chen; Matthew T. Weirauch; Gurjit K. Khurana Hershey; Hong Ji; Nasal DNA methylation differentiates corticosteroid treatment response in pediatric asthma: A pilot study. PLOS ONE 2017, 12, e0186150, 10.1371/journal.pone.0186150.
- Alvin T. Kho; Michael McGeachie; Kip G. Moore; Jody M. Sylvia; Scott Weiss; Kelan G. Tantisira; Circulating microRNAs and prediction of asthma exacerbation in childhood asthma. Respiratory Research 2018, 19, 128, 10.1186/s12931-018-0828-6.
- Alvin T. Kho; Sunita Sharma; Joshua S. Davis; Joseph Spina; Dagnie Howard; Kevin McEnroy; Kip Moore; Jody Sylvia; Weiliang Qiu; Scott T. Weiss; Kelan G. Tantisira; Circulating MicroRNAs: Association with Lung Function in Asthma. PLOS ONE 2016, 11, e0157998, 10.1371/journal.pone.0157998.
- Yu-Liang Yeh; M.-W. Su; B.-L. Chiang; Y.-H. Yang; C.-H. Tsai; Yungling L. Lee; Genetic profiles of transcriptomic clusters of childhood asthma determine specific severe subtype. Clinical Experimental Allergy 2018, 48, 1164-1172, 10.1111/cea.13175.
- Weiliang Qiu; Feng Guo; Kimberly Glass; Guo-Cheng Yuan; John Quackenbush; Xiaobo Zhou; Kelan G. Tantisira; Differential connectivity of gene regulatory networks distinguishes corticosteroid response in asthma. Journal of Allergy and Clinical Immunology 2018, 141, 1250-1258, 10.1016/j.jaci.2017.05.052.
- Michael T. Beste; Nicole Pfäffle-Doyle; Emily A. Prentice; Stephanie N. Morris; Uglas A. Lauffenburger; Keith B. Isaacson; Linda G. Griffith; Molecular Network Analysis of Endometriosis Reveals a Role for c-Jun-Regulated Macrophage Activation. Science Translational Medicine 2014, 6, 222ra16, 10.1126/scitranslmed.3007988.
- Michael J. McGeachie; George L. Clemmer; Boris Hayete; Heming Xing; Karl Runge; Ann Chen Wu; Xiaofeng Jiang; Quan Lü; Bruce Church; Iya Khalil; Kelan Tantisira; Scott Weiss; Systems biology and in vitro validation identifies family with sequence similarity 129 member A (FAM129A) as an asthma steroid response modulator. Journal of Allergy and Clinical Immunology 2018, 142, 1479-1488.e12, 10.1016/j.jaci.2017.11.059.
- Shiqi Tang; Jianwen Wang; Jishi Liu; Yan Huang; Yueyi Zhou; Shikun Yang; Wei Zhang; Minghui Yang; Hao Zhang; Niban protein regulates apoptosis in HK-2 cells via caspase-dependent pathway. Renal Failure 2019, 41, 455-466, 10.1080/0886022x.2019.1619582.
- Shintaro Katayama; Katarina Stenberg Hammar; Kaarel Krjutškov; Elisabet Einarsdottir; Gunilla Hedlin; Juha Kere; Cilla Soderhall; Acute wheeze-specific gene module shows correlation with vitamin D and asthma medication. European Respiratory Journal 2019, 55, 1901330, 10.1183/13993003.01330-2019.
- Peter Langfelder; Steve Horvath; WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559, 10.1186/1471-2105-9-559.
- Qiaoshi Lian; Bing Sun; Interferons command Trim22 to fight against viruses. Cellular & Molecular Immunology 2017, 14, 794-796, 10.1038/cmi.2017.76.
- Pité, H.; Morais-Almeida, M.; Rocha, S.M. Metabolomics in Asthma: Where Do We Stand? Curr. Opin. Pulm. Med. 2018, 24, 94–103.
- Anne H. Neerincx; Susanne J. Vijverberg; Lieuwe D. Bos; Paul Brinkman; Marc P. Van Der Schee; Rianne De Vries; Peter J. Sterk; Anke H. Maitland-Van Der Zee; Breathomics from exhaled volatile organic compounds in pediatric asthma. Pediatric Pulmonology 2017, 52, 1616-1627, 10.1002/ppul.23785.
- Su Jung Kim; Su Hee Kim; Ji Hyun Kim; Shin Hwang; Hyun Ju Yoo; Understanding Metabolomics in Biomedical Research. Endocrinology and Metabolism 2016, 31, 7-16, 10.3803/EnM.2016.31.1.7.
- Rachel Kelly; Joanne E. Sordillo; Sharon Lutz; Lydiana Avila; Manuel Soto-Quiros; Juan C. Celedón; Michael McGeachie; Amber Dahlin; Kelan G Tantisira; Mengna Huang; Clary B. Clish; Scott Weiss; Jessica Lasky-Su; Ann Chen Wu; Pharmacometabolomics of Bronchodilator Response in Asthma and the Role of Age-Metabolite Interactions.. Metabolites 2019, 9, 179, 10.3390/metabo9090179.
- Rachel Kelly; Bo L. Chawes; Kevin Blighe; Yamini V. Virkud; Damien Croteau-Chonka; Michael J. McGeachie; Clary B. Clish; Kevin Bullock; Juan C. Celedón; Scott Weiss; Jessica Lasky-Su; An Integrative Transcriptomic and Metabolomic Study of Lung Function in Children With Asthma. Chest 2018, 154, 335-348, 10.1016/j.chest.2018.05.038.
- Tomas Paulenda; Petr Draber; The role of ORMDL proteins, guardians of cellular sphingolipids, in asthma. Allergy 2016, 71, 918-930, 10.1111/all.12877.
- Antoaneta Toncheva; Daniel P. Potaczek; Michaela Schedel; Søren W. Gersting; Sven Michel; Natalie Krajnov; Vincent Gaertner; Julian M. Klingbeil; Thomas Illig; Andre Franke; Carla Winkler; Jens M. Hohlfeld; Christian Vogelberg; Andrea Von Berg; Albrecht Bufe; Andrea Heinzmann; Otto Laub; Ernst Rietschel; Burkhard Simma; Jon Genuneit; Ania C. Muntau; Michael Kabesch; Childhood asthma is associated with mutations and gene expression differences ofORMDLgenes that can interact. Allergy 2015, 70, 1288-1299, 10.1111/all.12652.
- Jennie G. Ono; Tilla S. Worgall; Stefan Worgall; 17q21 locus and ORMDL3: an increased risk for childhood asthma. Pediatric Research 2014, 75, 165-170, 10.1038/pr.2013.186.
- George Weinstock; Genomic approaches to studying the human microbiota. Nature 2012, 489, 250-256, 10.1038/nature11553.
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L; Analysis of the Microbiome: Advantages of Whole Genome Shotgun versus 16S Amplicon Sequencing. Biochem. Biophys. Biochemical and Biophysical Research Communications 2016, 469, 967-977, 10.1016/j.bbrc.2015.12.083.
- Yanjiao Zhou; Daniel Jackson; Leonard B. Bacharier; David Mauger; Homer Boushey; Mario Castro; Juliana Durack; Yvonne Huang; Robert F. Lemanske Jr; Gregory A. Storch; et al.George WeinstockKristine WylieRonina CovarAnne M. FitzpatrickWanda PhipatanakulRachel G. RobisonAvraham Beigelman The upper-airway microbiota and loss of asthma control among asthmatic children. Nature Communications 2019, 10, 5714, 10.1038/s41467-019-13698-x.
- Juliana Durack; Yvonne J Huang; Snehal Nariya; Laura S. Christian; K. Mark Ansel; Avraham Beigelman; Mario Castro; Anne-Marie Dyer; Elliot Israel; Monica Kraft; Richard J. Martin; David T. Mauger; Sharon R. Rosenberg; Tonya S. King; Steven R. White; Loren C. Denlinger; Fernando Holguin; Stephen C. Lazarus; Njira Lugogo; Stephen P. Peters; Lewis J. Smith; Michael E. Wechsler; Susan V. Lynch; Homer A. Boushey; National Heart, Lung and Blood Institute’s “AsthmaNet”; Bacterial biogeography of adult airways in atopic asthma.. Microbiome 2018, 6, 104, 10.1186/s40168-018-0487-3.
- Chung, K.F. Airway Microbial Dysbiosis in Asthmatic Patients: A Target for Prevention and Treatment? J. Allergy Clin. Immunol. 2017, 139, 1071–1081.
- Martin Depner; Markus J. Ege; Michael J Cox; Sarah Dwyer; Alan W. Walker; Lena T. Birzele; Jon Genuneit; Elisabeth Horak; Charlotte Braun-Fahrländer; Hanna Danielewicz; Raina M. Maier; Miriam F. Moffatt; William Osmond Cookson; Dick J.J. Heederik; Erika Von Mutius; Antje Legatzki; Miriam F. Moffat; Bacterial microbiota of the upper respiratory tract and childhood asthma. Journal of Allergy and Clinical Immunology 2017, 139, 826-834.e13, 10.1016/j.jaci.2016.05.050.
- Kathryn McCauley; Juliana Durack; Ricardo Valladares; Uglas W. Fadrosh; Din L. Lin; Agustin Calatroni; Petra K. Lebeau; Hoang Tran; Kei E. Fujimura; Brandon LaMere; Geil Merana; Kole Lynch; Robyn T. Cohen; Jacqueline Pongracic; Gurjit K. Khurana Hershey; Carolyn M. Kercsmar; Michelle Gill; Andrew H. Liu; Haejin Kim; Meyer Kattan; Stephen J. Teach; Alkis Togias; Homer A. Boushey; James E. Gern; Daniel J. Jackson; Susan V. Lynch; National Institute of Allergy and Infectious Diseases–sponsored Inner-City Asthma Consortium; Distinct nasal airway bacterial microbiotas differentially relate to exacerbation in pediatric patients with asthma. Journal of Allergy and Clinical Immunology 2019, 144, 1187-1197, 10.1016/j.jaci.2019.05.035.
- Yanjiao Zhou; Hongyu Gao; Kathie Mihindukulasuriya; Patricio S La Rosa; Kristine M Wylie; Tatiana A. Vishnivetskaya; Mircea L. Podar; Barbara B. Warner; Phillip I. Tarr; David E. Nelson; et al.J. Dennis FortenberryMartin J HollandSarah E BurrWilliam D ShannonErica SodergrenGeorge Weinstock Biogeography of the ecosystems of the healthy human body. Genome Biology 2013, 14, R1, 10.1186/gb-2013-14-1-r1.