Bone material strength is determined by several factors, such as bone mass, matrix composition, mineralization, architecture and shape. From a clinical perspective, bone fragility is classified as primary (i.e., genetic and rare) or secondary (i.e., acquired and common) osteoporosis
- bone fragility
- type I collagen
- post-translational modifications
1. Introduction
Developing bones consist of cartilaginous joints, the epiphysis, the growth plate cartilage with adjacent osteogenesis and the cortical and cancellous bone mineralized structure. Bone tissue contains three distinct cell types: (i) the osteoblasts, derived from mesenchymal cells, which deposit new bone tissue; (ii) osteoclasts, derived from bone marrow hematopoietic precursor cells, which break down bone matrix; and (iii) osteocytes (former osteoblasts) which orchestrate the activity of osteoblasts and osteoclasts as a response to mechanical strain [1]. The extracellular matrix of bone tissue is composed of inorganic minerals, collagen, water, non-collagenous proteins and lipids. Bone fragility can originate from alterations in all these components, be it the genetic blueprint, mechanical loading, insufficient remodeling at old age, estrogen deficiency or chronic medical conditions affecting bone accrual, structure or composition.
Traditionally, bone fragility is understood as resulting from reduced bone mass, or from defects in bone matrix composition or mineralization. Medical research has unveiled many monogenic bone fragility conditions, yet their mechanisms of disease remain incompletely understood. In fact, they continue holding secrets which may open doors for drug developments in rare and common osteoporosis.
2. Genetic Causes of Bone Fragility
2.1. Primary Osteoporosis Affecting Collagen (Osteogenesis Imperfecta)
2.1.1. Clinical Symptoms and Classification
Recurrent limb and vertebral fractures are typical for patients with osteogenesis imperfecta (OI). OI, also known as brittle bone disease, is an inherited disorder of connective tissue that has a wide clinical and genetic heterogeneity. OI is a rare disorder, with an incidence of one in 10,000–20,000 births [2]. From a bone material perspective, OI is characterized by low bone mass and increased bone mineralization density, which causes brittleness (
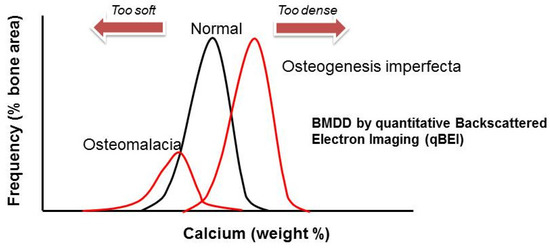
Figure 1.
2.1.2. Genetic Classification and Protein Function in OI
One recent classification [2] proposes five groups of subtypes for OI (Group A–E). Group A subtypes of OI (Type I–IV, XIII), which are caused by defects in collagen synthesis, structure or processing of COL1A1 and COL1A2, including their C-terminal propeptide cleavage by BMP1. Group B (type VII, VIII, IX and XIV) contains the genes that play a key role in post-translational modification of type I collagen, which are
CRTAP
LEPRE1
PPIB
TMEM38B
SERPINH1
FKBP10
PLOD2
P4HB
IFITM5
SERPINF1
SP7
WNT1
CREB3L1
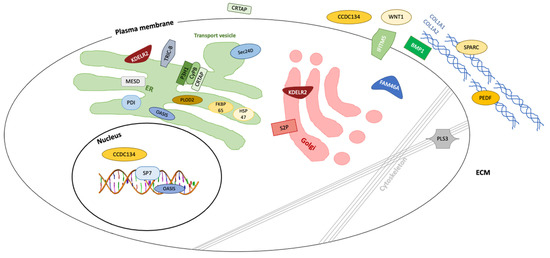
Figure 2.
All 24 OI-causing genetic entities have in common that they, by various mechanisms, affect the quality or quantity of type I collagen [2]. Since type I collagen is the main structure protein (94%) of pre-mineralized bone matrix (osteoid), abnormal type I collagen synthesis decreases bone mass and increases susceptibility to fracture. Type I collagen consists of three modified chains that form a triple helical fibril with two identical α1 (I) chains and one structurally similar but genetically different α2 (I) chain. The α chain is characterized by a strict pattern which contains a multiple triple sequence of a Gly (glycine)-X (normally proline)-Y (usually hydroxyproline). Notably, about a third of the residues are prolines that get hydroxylated. Each chain is characterized by amino- and carboxy-propeptides, which are critical in preventing self-assembly of collagen fibrils within the cells.
In the cells, type I collagen is synthesized as a soluble precursor molecule, procollagen with N-terminal and C-terminal propeptides that flank the helical domain. The biosynthesis of type I procollagen is a multistep process that involves an ensemble of proteins for post-translational modifications, folding, transport, secretion and quality control [3]. Procollagen synthesis is started up in the nucleus of collagen producing cells, such as osteoblasts and fibroblasts. The DNA segment is transcribed to precursor RNA that is spliced to mRNA and then transported to the endoplasmic reticulum (rER), via the N-terminal signal peptide, where it is translated to propolypeptide chains termed pro-α chains. In the rER, these propolypeptide chains undergo a series of post-translational modifications described in great detail elsewhere [3][4][5]. The C-terminal propeptide of each chain is attached to the rER membrane and folds into a structure that is stabilized by intra-chain disulfide bonds, which enables the selection and assembly of the correct chains into a triple helix.
2.1.3. Pathway-Specific Therapy
Neutralizing antibody against sclerostin (Scl-AB), a molecule produced by osteocytes to inhibit osteoblasts-mediated bone formation via the WNT signaling cascade, increased bone formation rate and bone mass in an OI preclinical mouse model [6].
Recently, preclinical studies demonstrated the potential of MSC transplantation before and after birth for severe types of OI (types II/III, severe type IV); this treatment approach is currently tested in the ongoing multicenter clinical trial boost brittle bones before birth (BOOSTB4) [7]. However, one major hurdle to such a cellular therapy is low engraftment of cells in all skeletal elements.
2.2. Primary Osteoporosis Caused by WNT-Signaling Pathway Defects
The Wingless-type mouse mammary tumor virus (MMTV) integration site family 1 (WNT1) protein belongs to a family of 19 secreted signaling glycoproteins. WNTs bind with the frizzled receptor and the co-receptors’ low-density lipoprotein receptor-related protein (LRP)-5 and -6 and activate the β catenin signal transduction pathway in various tissues, including bone [8][9]. This in turn leads to the translocation of β catenin into the nucleus, where it induces the expression of genes that regulate osteoblast differentiation [10]. Thus, the WNT signaling pathway controls mature osteoblast differentiation [11], bone development and bone maintenance [12]. Several groups have shown in patients from different countries that bi-allelic
WNT1 mutations are associated with moderate to severe cases of recessive OI type XV [13][14]. In addition, heterozygous pathogenic mutations in
WNT1
WNT1 bi-allelic nonsense, missense, splice site substitutions, deletion and frameshift mutations display severe bone fragility, reduced bone mass, multiple fractures and growth delay [14][15]. The structural bone phenotype includes low bone turnover with an imbalance between formation and resorption. This imbalance may be mediated through WNT1 function in osteocytes [16]. Similar to the human nonsense
WNT1
wnt1 mutation in exon 3 (c.565delG, p.Glu189Argfs*10) [13][17]. This mouse model shows typical OI features including severe osteopenia, spontaneous fractures, reduced bone strength and impaired matrix mineralization, mimicking the human disease. Notably, bone forming osteoblast function is impaired, while bone resorbing osteoclast function is not changed in this mouse model [18].
WNT1 mutations arrest the downstream intracellular signaling cascade and nuclear translocation of β catenin and the expression of the regulated genes [10][13]. Loss of function of β catenin results in osteochondroprogenitor cells differentiating into chondrocytes instead of osteoblasts. On the other hand, gain of function of WNT signaling result in enhancement of osteoblast differentiation in vitro [19]. In addition, the WNT signaling cascade in osteocytes plays a critical role in regulating bone cell homeostasis [20]. Mice deficient with β catenin in osteocytes show bone loss [20].
WNT1
LRP5 cause osteoporosis-pseudoglioma syndrome, which is characterized by severe bone fragility and ocular manifestations [21], and monoallelic
LRP5 mutations, which cause primary, autosomal dominant osteoporosis [22]. Hence, there appears to be a gene dosing effect in defective WNT signaling. Moreover, patients with gain of function and loss of function mutations in the LRP5 have high or low bone mass disorders resulting from constitutive activation or decreased osteoblast activity, respectively [14][23]. This fact further supports the notion of a gene dosing effect and makes elements of the WNT signaling pathway attractive drug targets. Trials have been conducted in adult osteoporosis, which led to the market approval of antibody therapy against sclerostin, an inhibitor of the WNT signaling pathway [24]. Trials in children are awaited.
2.3. Primary Osteoporosis Caused by Defects in the TGF-β Pathway
Polypeptides in the transforming growth factor β (TGF-β) family are involved in controlling cell activity and metabolism in bone and cartilage tissues. After TGF-β release from the ECM, it interacts with a receptor complex containing type I (TβRI, TGFBR1) and type II (TβRII, TGFBR2) subunits. The highly complex pathway involves intracellular signal transduction through cytoplasmic proteins, belonging to transcription factors from the SMAD family. A variety of heritable skeletal conditions is associated with dysregulated TGF-β signaling, including Camurati-Engelmann disease [MIM: 131300] [25][26][27][
67], Marfan syndrome [MIM: 154700] [26] and Loeys-Dietz syndrome [MIM: 613795] [27] but also OI [28]. Bone fragility is a distinct phenotypic feature of Loeys-Dietz syndrome, caused by mutations in SMAD3.
2.4. Primary Osteoporosis Caused by RANKL/RANK/OPG Defects: TNFRSF11B (Juvenile Paget Disease) and TNFRSF11A (Familial Expansile Osteolysis)
The Receptor Activator of Nuclear Factor Kappa B (RANK, TNFRSF11A) and its ligand RANKL play a key role in osteoclast activation and differentiation [29]. RANKL is a cytokine expressed by osteoblast lineage cells, including osteoblasts and osteocytes. RANKL binds with its cognate receptor RANK on the surface of osteoclast precursors, which activates cell differentiation. Hence, RANKL is essential for formation and activation of osteoclasts. Mice and humans lacking RANKL display complete abrogation of osteoclastogenesis. Osteoblast also express a decoy receptor osteoprotegerin (OPG, TNFRSF11B) that competitively binds with the RANK receptor and inhibits the interaction with RANKL. Hence, osteoblasts and osteocytes regulate activation and differentiation of osteoclasts by the RANKL-RANK axis signaling pathway [29][30].
TNFRSF11B encoding the decoy receptor OPG results in unopposed RANK activation and permanently elevated bone turnover, a condition called Juvenile Paget’s disease (JPD; [MIM: 602080]) [31]. JPD is a rare autosomal recessive disease which develops during infancy and early childhood, and worsens in adolescence with pain from debilitating fractures and deformities caused by highly accelerated bone turnover of the entire skeleton [32]. JPD also has extra-skeletal manifestations, including bowing deformities and fractures, contractures as well as short stature [33]. Patients demonstrate histopathological evidence of high bone turnover and weak, disorganized woven bone [34][35].
TNFRSF11A cause familial expansile osteolysis [FEO; MIM: 174810], leading to permanent activation of RANK. The ensuing progressive osteoclastic resorption is associated with medullar expansion and severe pain, disabling deformities and pathological fractures. Characteristically, FEO is accompanied by deafness and loss of dentition as a result of middle ear and jaw bone abnormalities, and variably raised serum alkaline phosphatase levels. FEO cases present with osteolytic lesions in long bones, whereas JPD patients tend to present with trunk and skull lesions [36].
2.5. Bone Fragility in Hajdu Cheney Syndrome
In bone, NOTCH signaling is involved in the regulation of bone formation and bone resorption [37][88]. In vitro studies have suggest that the NOTCH signaling cascade modulates signaling downstream of RANK, hence activating NOTCH2 enhances osteoclasts maturation [38][89]. NOTCH2 plays a key role in skeletal development [39][90]. Homozygous deletion of mouse Notch2 results in early embryonic lethality [40][91]. Heterozygous gain of function mutations in NOTCH2 lead to Hajdu-Cheney syndrome (HCS) [41][92]. HCS [MIM: 102500] is a rare autosomal dominant disease characterized by severe osteoporosis associated with craniofacial dysmorphism, acroosteolysis and Wormian bones [42][93]. Nonsense or short deletion mutations in NOTCH2 exon 34 (the last exon) result in an early termination upstream of the Proline-Glutamic acid-Serine-Threonine (PEST) domain, at the end of the protein, which is required for the NOTCH2 receptor ubiquitination and degradation [43][94]. Hence, these mutations produce a truncated protein lacking the proteolytic degradation domain of NOTCH2 receptors, leading to sustained NOTCH2 activation with increased osteoclastogenesis [44][95]. Histomorphometric analysis in affected individuals demonstrate increased bone resorption, increased heterogeneity of mineralization and woven bone. Typical features are reduced cortical thickness and low bone mass. Given the increased osteoclast numbers and turnover, affected individuals respond well to bisphosphonate therapy [45][96].