The exact connection between Alzheimer’s disease (AD) and type 2 diabetes is still in debate. However, poorly controlled blood sugar may increase the risk of developing Alzheimer’s. This relationship is so strong that some have called Alzheimer’s “diabetes of the brain” or “type 3 diabetes (T3D)”. Given more recent studies continue to indicate evidence linking T3D with AD, this state-of-the-art aimed to demonstrate the relationship between T3D and AD based on the fact that both the processing of amyloid-β (Aβ) precursor protein toxicity and the clearance of Aβ are attributed to impaired insulin signaling, and that insulin resistance mediates the dysregulation of bioenergetics and progress to AD.
- Alzheimer’s disease
- hypometabolism
- type 2 diabetes
- type 3 diabetes
- insulin resistance
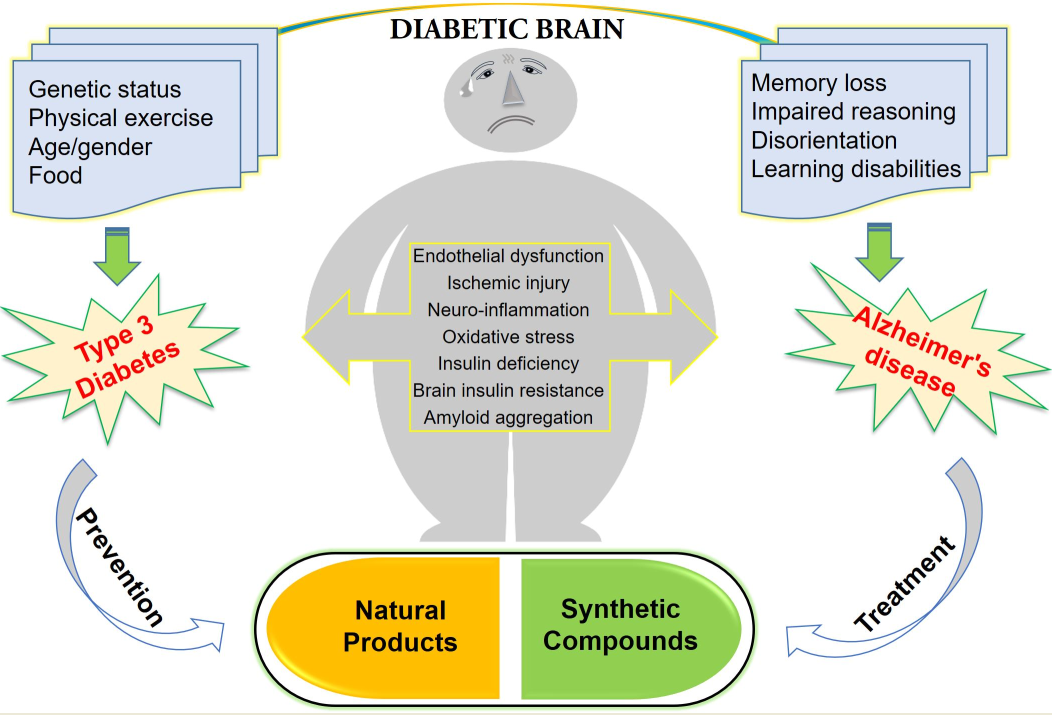
1. The Role of Type 3 Diabetes in Glucose Homeostasis
The key to understanding the relationship between diabetes and these other areas begins with the role of energy homeostasis in diabetes. Energy homeostasis is a well-regulated process that depends on the coordination between feeding behavior and energy expenditure. The control of energy homeostasis in humans has received much attention in recent years due to alterations caused by onset of conditions such as obesity and diabetes. There are two distinct features of adult neurons that make them vulnerable to either neuronal cell death or a diseased state such as neurodegeneration or neuronal loss. The first feature is that fully differentiated (adult) neurons are permanently postmitotic cells, which lack regenerative ability [1]. Therefore, when adult neurons are exposed to any cellular stresses such as lack of adenosine triphosphate (ATP) moieties or energy crisis or oxidative stress, they either die or experience apoptosis, or degenerate or cause neuronal degeneration and loss, and thus predispose neurodegenerative diseases [1]. The second important feature is that brain neurons or tissues are highly demanding excitable cells, in which more than 40% of the present ATP is used to keep neurons viable or alive [2]. There are two sources of brain glucose that involve cortical glucose metabolism stimulation through basal insulin levels [3] and astrocytic glycogen conversion to glucose that is stimulated by the activation of glial β-adrenoceptors. The increase in glucose uptake is transported by insulin-sensitive glial glucose transporter type 1 (GLUT1) to the plasma membrane for neuronal use. Therefore, the balanced cellular glucose transportation depends on astrocytes and glucose transporters that are expressed in the brain [4].
Moreover, a glucose homeostasis defect might be important in the pathogenesis of T3DM due to impaired glucose uptake as a result of impaired glucose metabolism in the brain. The mechanisms that are involved in glucose transportation abnormalities include brain insulin resistance and intracellular glucose metabolic disturbance. These two abnormalities may contribute to cerebral glucose hypometabolism in T3DM or the brain insulin resistance disease state. A decreased glucose transporters correlated to abnormal hyperphosphorylation of tau in neurodegenerative diseases was reported [5]. Therefore, impairment of insulin signaling not only affects systemic glucose blood levels but also causes various degenerative processes or neuronal cell death or loss [6]. In addition, insulin resistance in T2DM has been defined as “reduced sensitivity in body tissues to the action of insulin” [7]. Similarly, brain insulin resistance can be defined as the failure of brain cells to respond to insulin and its corresponding IRs [8]. Consequently, this leads to insulin deficiency and impaired glucose transport inside the neurons due decreased number of expressed GLUTs in the cell membrane. Furthermore, insulin resistance in the CNS correlates with insulin resistance in the periphery. Therefore, loss of responsiveness to insulin could make neurons more susceptible to neurotoxic insults due to their being devoid of protective effect of insulin [9]. Furthermore, insulin-resistant patients have many increased pathologic features such as apoptosis, neurodegeneration, and the resultant decline in cognition.
The desensitization of the neuronal insulin receptor in brain insulin resistance, similar to the process in T2DM, may play a key role in causing T3DM and its future complications [10]. Besides, T2DM is a metabolic syndrome characterized by insulin resistance, which is also a pathological feature of neurodegeneration or neuroendocrine disorder or T3DM [3]. Thus, glucose homeostasis plays a role in T3DM pathogenesis. Brain glucose uptake or metabolism is impaired in T3DM. Therefore, the combination of T2DM and neurodegenerative brain diseases may be considered as this new classification of diabetes, called T3DM or a neuroendocrine disorder.
2. Type 3 Diabetes and Aβ Protein Pathology
Amyloidosis is a pathological condition which consists of the accumulation of fibrillary proteins, characterizing by extracellular amyloid deposits with a clinical variability depending on the affected tissue. Recently, there has been newly emerged evidence regarding the relationship between the pathogenesis of AD and insulin resistance. It is important to consider T2DM as a risk factor essential for the formation of deposits of amyloid-β in patients’ brains with dementia. There was a toxic cycle between continuous insulin exposure and Aβ accumulation inside the neurons [11]. According to Farris et al., insulin degrading enzyme (IDE) regulates the levels of insulin, Aβ protein, and amyloid precursor protein (APP) intracellular domain in vivo [11]. This study showed that a rat model of T2DM of mutant IDE was associated with hyperinsulinemia and glucose intolerance, as hallmarks of T2DM and T3DM or brain insulin resistance. This implies that IDE hypofunction may underlie or contribute to some forms of T3DM and T2DM and provide a mechanism for the recently recognized association among hyperinsulinemia, diabetes, and neurodegeneration or neuronal loss [11]. Therefore, in normal subjects, IDE reduces Aβ, regulates insulin and also degrades APP intracellular domain (AICD). Thus, there was a regulatory relationship among insulin, IDE and Aβ. In the case of brain insulin resistance, insulin possibly failed to stimulate the clearance of Aβ, which permits its buildup inside the neurons causing neurodegeneration or neuronal loss, as hallmarks of T3DM or brain insulin resistance [11]. There is a debate about T3DM and brain insulin resistance as to whether it is a consequence or a cause of abnormal Aβ expression and protein processing [12]. In terms of the concept of T3DM being a consequence, Aβ toxicity may cause insulin resistance in the brain. The Aβ disturbs insulin signaling by competing with insulin on its receptors [13], reducing the surface expression of IRs, and reducing the insulin affinity to its relative receptors, and interfering directly with phosphatidylinositol-4, 5- bisphosphate 3-kinase (PI3K)/Akt activation, causing a blockade of its signaling and leading to impaired survival signaling, increased activation of GSK-3β activity, and increased hyperphosphorylation of tau [14].
On the other hand, in terms of the concept of T3DM being the cause, the brain insulin resistance with oxidative stress and neuroinflammation may cause Aβ accumulation, as shown in Figure 12. The studies that incorporate this concept claim that insulin stimulation may increase or accelerate trafficking of Aβ from the Golgi network to the plasma membrane. Therefore, insulin may activate Aβ extracellular excretion and, at the same time, inhibit its intracellular accumulation by activating its degradation by the insulin-degrading enzyme (IDE) [15]. Thus, impaired insulin signaling can disturb both APP processing and Aβ clearance [16]. This leads to increased neurotoxic effects of Aβ on neurons, resulting in possible neurodegeneration and neuronal cell death. T2DM and AD patients have similar amyloid beta deposits both in pancreas and in the brain. Several researchers have suggested this new pathology should be addressed as T3D [17][18][19][20]. Some of the target receptors of T2DM such as the insulin-like growth factor 1 (IGF-1) and peroxisome proliferator-activated receptor gamma (PPARG) are also involved in the regulation of the expression and phosphorylation of tau protein [20].
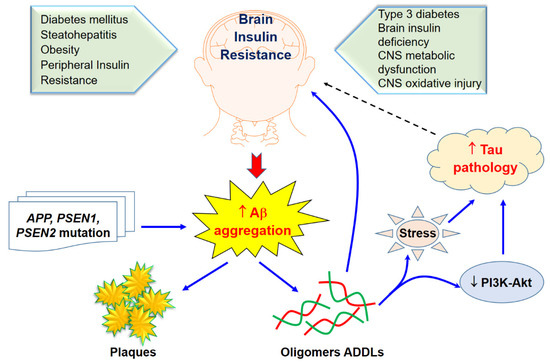
Figure 12. Brain insulin resistance and Aβ aggregation and its toxicity. Solid arrows indicate the interactions of Aβ aggregation on brain insulin resistance through sone potential pathways while tau pathology would likely effect of brain insulin as revealed in a dasher arrow.
3. Type 3 Diabetes Regarding Alzheimer’s Disease
Insulin resistance in AD and diabetes can lead to hyperinsulinemia, thereby, saturating insulin-degrading enzymes (IDE) for insulin and Aβ degradation. Recently, many studies indicated that the incidence of AD is higher in T2D patients and obese individuals, implying common mechanisms driving these disorders [21][22][23]. Insulin resistance could be a main feature which is shared among diabetes, obesity, and AD [24]. The neuronal glucose uptake may not depend on insulin totally, thus the concept of insulin resistance in the brain is more related to impaired insulin signaling pathways. The malfunction of insulin signaling pathways and resultant state of hypometabolism observed are considering among factors in altered bioenergetics that connects AD and T2D [25]. The insulin resistant state could lead to compromised neuron functions and cognitive skills, accompanied by an extreme rise in insulin and relatively declined insulin activity in the periphery as important predictors of T2D [26][27]. Consequently, this leads to the development of neuritic plaques, hippocampal atrophy, cognitive performance and lower cerebrocortical glucose metabolism which may closely correlate with memory impairments [19]. A previous study revealed that increased p-Ser312IRS1 manifested in prodromal AD patients that sustained these alterations a decade ago as AD patients [28], suggesting that insulin resistance in AD develops years before clinical manifestations and that neural-derived exosomes carry potential for early AD diagnosis. Due to lack of insulin response, down regulation of insulin receptors, reduced binding of insulin receptors or faulty activation of the insulin signaling cascade cause the defective brain insulin signaling in AD and T2D. The major consequence of this altered cascade is the decreased neuronal glucose uptake that is manifested as impaired neuroplasticity, neurotransmitter deficits, collapse of bioenergetics mechanism and initiation of fateful inflammatory cascade. Overall, the consequences of impaired insulin signaling are attributed to impaired metabolism in the brain that may lead to brain malfunction, providing possible explanations for the connection between diabetes, obesity, and AD [29], as shown in Table 1.
Table 1.
Causal model for the potential associated with between T3D and AD.
| Upstream Risk Factors | Metabolic Precursors | Pathways | Subclinical Pathology | Disease Outcome | |
|---|---|---|---|---|---|
| Social factors: stress, low socioeconomic status, certain ethnic and racial groups | Obesity Visceral Adiposity |
Vascular Processes | Blood pressure and hypertension Hyperlipidemia Apolipoprotein E |
Cerebral blood flow Atherosclerosis |
Amyloid precursor proteins | Alzheimer’s disease |
| Poor diet: high in calories, fat and sugar, low in fiver | Inflammatory/Oxidative processes | Inflammation Oxidative stress Endothelial function |
Neurofibrillary Tangles Amyloid B deposits |
|||
| Physical inactivity Genetics and family history |
Hyperglycemia Hyperinsulinemia |
Metabolic processes | Insulin resistance Insulin-degrading enzyme Peroxisome proliferative-activated receptors |
|||
| Early childhood exposures in utero and birth weight | Brain and hippocampal atrophy White matter hyperintensities |
|||||
Insulin resistance or dysfunction of insulin signaling is a universal feature of T2D, due to altered glucose metabolism and its interdependence on cell death pathways form the basis of linking T3D with AD, as shown in Figure 23. T3D occurs when neurons in the brain become unable to respond to insulin, which is essential for basic tasks, including memory and learning. Some researchers believe insulin deficiency is central to the cognitive decline of AD. Dysfunctional insulin pathways and resistance of insulin is a status of receptor dysfunction, altered receptor expression, deviations in receptor binding and malfunctioned events in the phosphorylation chain or the altered activities related to kinases involved in phosphorylation. At the molecular level, a cell senses insulin through insulin receptors, with the signal propagating through a signaling cascade collectively known as the PI3K/Akt/mTOR signaling pathway. Recent studies suggested that the pathway operates as a bistable switch under physiologic conditions for certain types of cells, and insulin response may well be a threshold phenomenon [30][31][32]. The pathway’s sensitivity to insulin may be blunted by many factors such as free fatty acids, causing insulin resistance. It also is based on the finding that insulin resistance may be reversed rapidly by exposing cells to mitochondrial uncouplers, electron transport chain inhibitors, or mitochondrial superoxide dismutase mimetics [33][34].
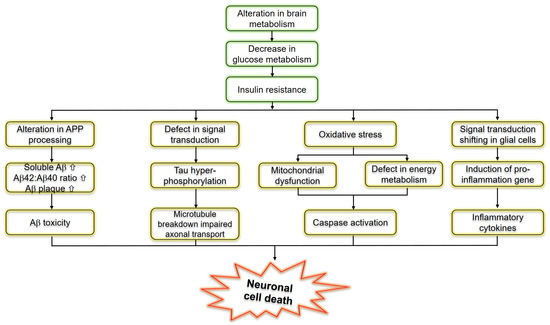
Figure 23.
Schematic representation of molecular pathways linking insulin resistance and Alzheimer’s disease.
Interestingly, impaired insulin signaling is present in several transgenic and nontransgenic mouse models of AD. Some previous clinical studies have reported that AD patients could have glucose intolerance, suggesting a bidirectional relationship between the two conditions [35][36]. Reduced levels of IRS-1 associated to the membrane of hippocampal extracts [37] and a decreased activation of IRS-1 and PI3K in the hippocampus and cortex were observed in ten-month-old mice [38]. Markers of insulin resistance were also reported in the hypothalamus of APP/PS1 mice [39] since the IRS-1 phosphorylated in serine 616 in the hippocampus at nine months of age was higher than that of the control group [40], and increased levels of IRS-1 phosphorylated in serine 636 and 312 in the frontal cortex at 13 months [41] were also demonstrated. In combination with peripheral insulin resistance, there was also a report of an increased inhibitory phosphorylation of IRS-1 in serine 612 in the hippocampus of five-month-old tg2576 mice [38]. Remarkably, the central infusion of AβOs lead to peripheral insulin resistance, which was further observed in the APP/PS1 and in the 3xTgAD mouse models of AD [42]. To confirm these concepts, further evidence is still required to investigate the mechanisms whereby AD affects the diabetic phenotype. T3D regarding AD and its approaches for treatment and prevention using naturally synthetic compounds, as shown in Figure 12.
In addition, in the wake of the worldwide increase in T2DM, a major focus of research aims to understand the signaling pathways impacting this disease. Insulin signaling regulates glucose, lipid, and energy homeostasis, predominantly via action on liver, skeletal muscle, and adipose tissue. Cell signaling pathways can be described by a list of biomolecular reactions which occur between the pathway components. T2DM associated with impaired insulin and insulin-like growth factor-1 (IGF1) signaling (IIS) is a risk factor for cognitive impairment and dementia including AD [43]. Importantly, systemic heterozygous inactivation of IGF1R (IGF1R+/−) or neuronal deletion of IGF1R (nIGF1R−/−) could improve the survival in the Tg2576 mouse model of AD while reducing behavioral impairment and Aβ accumulation [44]. Reduced IRS2 signaling throughout the body or in the brain prolongs life span [45] may lead to systemic reduction of IRS2 (IRS2−/−), improves cognitive function, and reduces Aβ deposition and premature mortality in Tg2576 mice with normal blood glucose levels [44][46]. Hence, more recent animal studies have revealed that a reduction in intracellular signaling mediated by IGF1R-IRS2 signaling but not the IR cascade in the CNS exerts neuroprotective effects in AD animal models [43].
4. Therapeutic Approaches to Type 3 Diabetes in Alzheimer’s Disease
Insulin resistance is well known as an essential feature of T3D, therefore treatment strategies for T3D, particularly those aimed at improving insulin sensitivity, may also benefit those patients at risk for AD at the early stages. Due to the overlapping yet distinct pathological features among diabetes, insulin resistance and cognitive decline, multitargeted drug therapies along with lifestyle interventions are also explored [47] from the perspective of research in the pharmaceutical industry, including nutraceuticals, antioxidant activity [48], polyphenols, omega-3 fatty acids as well as the brain–gut connections [49].
Among nutraceuticals produce, a brain-permeable compound, curcumin is able to target abnormal protein aggregates [50]. Curcumin may also thwart “proapoptotic signaling pathways in primary hippocampal neuron cultures”. Previous research has also shown the benefit of metformin in mice when coupled with curcumin and piperine supplementation, particularly regarding enhanced insulin sensitivity, signaling, and better systemic glucose tolerance [50], promising natural substances for AD patients. However, the anti-inflammatory benefits of fruits and vegetables have been widely publicized for decades, particularly regarding antioxidant action in reducing inflammatory damage [51]. Rodent research has linked various vegetables and fruits as protective “against cognitive and brain neuropathology from dietary oxidative stress” due to innumerable bioactive constituents such as carotenoids, antioxidant vitamins, polyphenols and flavonoids [52]. A various families of flavonoids have been suggested to be the potential therapeutic implications via in vivo models [53]. This has significant potential to advance our understanding of proactive approaches toward preventing AD and inhibiting progression. The essential role of omega-3 fatty acids in brain development and maintenance has been well recognized, particularly in the past ten years, yet only recently “have their effects on brain aging been explored” [54]. Diets rich in omega-3 fatty acids and naturally low in omega-6fatty acids may hold the key for nutritional therapy for AD patients [55]. The ketogenic diet may even diminish and clear beta amyloid plaques within the brain, while convalescing damaged mitochondria and reducing universal inflammation [56]. New research has shown that glycated APOE4 protein and faulty insulin signaling leads not only to impaired energy transport for brain tissues, but also impaired lipid transportation, mainly cholesterol [56][57]. APOE4 accounted for approximately 20% of the general population and >50% among Alzheimer’s cases, is responsible for interrupting how the brain processes insulin [58]. The gene and the peripheral insulin resistance caused by the high-fat diet together induced insulin resistance in the brain [59]. The APOE4 protein produced by the gene can bind more aggressively to insulin receptors on the surfaces of neurons than its normal counterpart, APOE3. APOE4 goes on to do lasting damage to brain cells. After blocking the receptor, the sticky APOE4 protein begins to clump and becomes toxic [59]. Furthermore, once the protein enters the interior of the neuron, the clumps get trapped within the cell’s machinery, impeding the receptors from returning to the neuron surface to do their work. The insulin signal processing gets increasingly more impaired, starving brain cells. There is no pharmaceutical intervention that has ever existed that has been more potent in improving overall vasculature throughout the body, than exercise [60]. This also has extensive implications for AD patients and type 2 diabetics due to increases in quality of life, neurochemical messaging within the brain, restorative power over insulin resistance, and the ability to clear Aβ plaques in certain individuals [60]. The concept of the gut–brain axis, the bidirectional communication between gut and brain, contributing significantly to the pathogenesis of AD that has been supported by many experimental and clinical studies [49]. Representatives of some compounds and drugs for the treatment or prevention of T3D regarding AD progression are presented in Table 2.
Table 2. Summary of representative of preclinical and clinical studies on the efficacy of antidiabetic, insulin-sensitizing drugs on multiple aspects of AD pathology.
| Compound | Potential Pathway | Study Design | Reference | |
|---|---|---|---|---|
| DA5-CH | Reduces tau phosphorylation and normalizes theta rhythm | Injected intracerebroventricula (ICV), streptozotocin on rat | [61] | |
| DA-JC1 | Antagonizing circadian rhythm disorders induced by Aβ | 31–35 | ICV, amyloid(31–35) AD model | [62] |
| DA5-CH | Improved of hippocampal synaptic plasticity and activation of the PI3K/AKT signaling pathway | APP/PS1 mouse model of AD | [63] | |
| DA-CH3 | Reduced ER stress and apoptotic signaling, reduced amyloid plaque load in the brain | APP/PS1 mouse model of AD | [64] | |
| Insulin | Prevention of Aβ oligomer induced synapse loss and insulin receptor reduction, amelioration of PKR-mediated ER stress | Rat hippocampal neuronal cultures | [65][66] | |
| Insulin | AD patients that are not ε4 carriers have reduced sensitivity to insulin, effecting cognitive performance | AD patients homozygous or not for the ApoE-ε4 allele and normal subjects intravenously injected | [67] | |
| Insulin | Improved verbal memory in MCI AD ε4-subjects after acute insulin administration, but not in ε4 carriers | AD patients homozygous or not for the ApoE-ε4 allele, MCI patients and most subjects intranasally administrated | [68][69] | |
| Insulin | Chromic intranasal insulin doses enhanced selective attention, retention of new information and functional status of MCI and early AD subjects | AD patients, MCI patients and normal subjects intranasally administrated | [70] | |
| Insulin | Only women presented improved working memory after treatment | Healthy men and woman intranasally administrated | [71] | |
| Liraglutide | Reduction of tau phosphorylation; protection of insulin reception and synapse loss in a c-AMP dependent manner | Cynomolgus monkeys ICV with Aβ oligomer | [72] | |
| Liraglutide | Improvement of memory deficits in novel object recognition test and fear conditioning | Swiss mice injected ICV with Aβ oligomer | [72] | |
| Liraglutide | Restored memory deficits in object recognition test and Morris water maze; enhanced LTP; reduced microglial activation; diminished Aβ plaque load | APP/PSEN1 mice | [73][74] | |
| Exendin-4 | Decrease in the inhibitory phosphorylation of Ser312IRS1, Ser66IRS1 of INK, while restoring activating Tyr465 IRS1 phosphorylation | Rat hippocampal neural cultures | [41] | |
| Exendin-4 | Improvement of spatial memory in the Morris water maze; reduced amyloid plaque LOAD | APP/PS1 mice | [41] | |
| Exedin4- Liraglutide | eIF2α phosphorylation reduction | Rat hippocampal neural cultures, APP/PS1 mice, cynomolgus monkeys injected ICV with Aβ oligomer | [66] | |
| GLP-1 Exendin-4 | Reduction of neural excitotoxicity | Rat hippocampal neural cultures, rats injected on the basal nucleus with ibotenic acid | [75] | |
| Rosiglitazone | Reversal of memory deficits in objects recognition test and the Morris water maze; Aβ levels reduction | AD transgenic mice J20 line | [76] |
References
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378.
- Paolo Gubellini; Barbara Picconi; Massimiliano Di Filippo; Paolo Calabresi; Downstream mechanisms triggered by mitochondrial dysfunction in the basal ganglia: From experimental models to neurodegenerative diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2010, 1802, 151-161, 10.1016/j.bbadis.2009.08.001.
- J Apelt; G Mehlhorn; R Schliebs; Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. Journal of Neuroscience Research 1999, 57, 693–705.
- Zhichun Chen; Chunjiu Zhong; Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Progress in Neurobiology 2013, 108, 21-43, 10.1016/j.pneurobio.2013.06.004.
- Ying Liu; Fei Liu; Khalid Iqbal; Inge Grundke-Iqbal; Cheng-Xin Gong; Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Letters 2008, 582, 359-364, 10.1016/j.febslet.2007.12.035.
- Li, L.; Holscher, C; Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402.
- Barry J Goldstein; Insulin resistance as the core defect in type 2 diabetes mellitus. The American Journal of Cardiology 2002, 90, 3g–10g, 10.1016/s0002-9149(02)02553-5.
- John G. Mielke; Changiz Taghibiglou; Lidong Liu; Yu Zhang; ZhengPing Jia; Khosrow Adeli; Yu Tian Wang; A biochemical and functional characterization of diet-induced brain insulin resistance. Journal of Neurochemistry 2005, 93, 1568-1578, 10.1111/j.1471-4159.2005.03155.x.
- Trevor Hardigan; Rebecca Ward; Adviye Ergul; Cerebrovascular complications of diabetes: focus on cognitive dysfunction. Clinical Science 2016, 130, 1807-1822, 10.1042/cs20160397.
- S. Hoyer; The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: an update. Journal of Neural Transmission 2002, 109, 341-360, 10.1007/s007020200028.
- Wesley Farris; Stefan Mansourian; Yang Chang; Loren Lindsley; Elizabeth Eckman; Matthew P. Frosch; Christopher B. Eckman; Rudolph E. Tanzi; Dennis J. Selkoe; Suzanne Y. Guénette; Insulin-degrading enzyme regulates the levels of insulin, amyloid -protein, and the -amyloid precursor protein intracellular domain in vivo. Proceedings of the National Academy of Sciences 2003, 100, 4162-4167, 10.1073/pnas.0230450100.
- Suzanne M. De La Monte; Brain insulin resistance and deficiency as therapeutic targets in Alzheimer's disease. Current Alzheimer Research 2012, 9, 35-66, 10.2174/156720512799015037.
- Xie Ling; Ralph N. Martins; Marco Racchi; S. Craft; E. Helmerhorst; Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. Journal of Alzheimer's Disease 2002, 4, 369-374, 10.3233/jad-2002-4504.
- Wen-Hua Zheng; Satyabrata Kar; Remi Quirion; Insulin-like Growth Factor-1-induced Phosphorylation of the Forkhead Family Transcription Factor FKHRL1 Is Mediated by Akt Kinase in PC12 Cells. Journal of Biological Chemistry 2000, 275, 39152-39158, 10.1074/jbc.m002417200.
- Laura Gasparini; Gunnar K. Gouras; Rong Wang; Rachel S. Gross; M. Flint Beal; Paul Greengard; Huaxi Xu; Stimulation of β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. The Journal of Neuroscience 2001, 21, 2561-2570, 10.1523/JNEUROSCI.21-08-02561.2001.
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease, Alzheimer’s & Dementia: Diagnosis. Assess. Dis. Monit. 2019, 11, 392–404.
- Khyati Mittal; Ruchi Jakhmola Mani; Deepshikha P. Katare; Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Scientific Reports 2016, 6, 25589, 10.1038/srep25589.
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. A J. Clin. Ther. 2009, 14, 373–379.
- Konrad Talbot; Hoau-Yan Wang; Hala Kazi; Li-Ying Han; Kalindi P. Bakshi; Andres Stucky; Robert L. Fuino; Krista R. Kawaguchi; Andrew J. Samoyedny; Robert S. Wilson; Zoe Arvanitakis; Julie A. Schneider; Bryan A. Wolf; David A. Bennett; John Q. Trojanowski; Steven E. Arnold; Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline.. Journal of Clinical Investigation 2012, 122, 1316-38, 10.1172/JCI59903.
- Suzanne M. De La Monte; Type 3 diabetes is sporadic Alzheimer׳s disease: mini-review. European Neuropsychopharmacology 2014, 24, 1954-1960, 10.1016/j.euroneuro.2014.06.008.
- Laura D. Baker; Donna Cross; Satoshi Minoshima; Dana Belongia; G. Stennis Watson; Suzanne Craft; Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults With Prediabetes or Early Type 2 Diabetes. Archives of Neurology 2011, 68, 51-57, 10.1001/archneurol.2010.225.
- Miia Kivipelto; Tiia Ngandu; Laura Fratiglioni; Matti Viitanen; Ingemar Kåreholt; Bengt Winblad; Eeva-Liisa Helkala; Jaakko Tuomilehto; Hilkka Soininen; Aulikki Nissinen; Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Archives of Neurology 2005, 62, 1556-1560, 10.1001/archneur.62.10.1556.
- George Razay; Anthea Vreugdenhil; Gordon Wilcock; Obesity, Abdominal Obesity and Alzheimer Disease. Dementia and Geriatric Cognitive Disorders 2006, 22, 173-176, 10.1159/000094586.
- Stephanie Kullmann; Martin Heni; Manfred Hallschmid; Andreas Fritsche; Hubert Preissl; Hans-Ulrich Häring; Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiological Reviews 2016, 96, 1169-1209, 10.1152/physrev.00032.2015.
- Sami Gabbouj; Simo Ryhänen; Mikael Marttinen; Rebekka Wittrahm; Mari Takalo; Susanna Kemppainen; Henna Martiskainen; Heikki Tanila; Annakaisa Haapasalo; Mikko Hiltunen; et al.Teemu Natunen Altered Insulin Signaling in Alzheimer's Disease Brain - Special Emphasis on PI3K-Akt Pathway. Frontiers in Neuroscience 2019, 13, 629, 10.3389/fnins.2019.00629.
- Stephen Lillioja; David M. Mott; Maximilian Spraul; Robert Ferraro; James E. Foley; Eric Ravussin; William C. Knowler; Peter H. Bennett; Clifton Bogardus; Insulin Resistance and Insulin Secretory Dysfunction as Precursors of Non-Insulin-Dependent Diabetes Mellitus: Prospective Studies of Pima Indians. New England Journal of Medicine 1993, 329, 1988-1992, 10.1056/NEJM199312303292703.
- Jun Li; Litao Bai; Fan Wei; Jing Zhao; Danwei Wang; Yao Xiao; Weitian Yan; Junping Wei; Therapeutic Mechanisms of Herbal Medicines Against Insulin Resistance: A Review. Frontiers in Pharmacology 2019, 10, 661, 10.3389/fphar.2019.00661.
- Dimitrios Kapogiannis; Adam Boxer; Janice B. Schwartz; Erin L. Abner; Arya Biragyn; Umesh Masharani; Lynda Frassetto; Ronald C. Petersen; Bruce L. Miller; Edward J Goetzl; et al. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural‐derived blood exosomes of preclinical Alzheimer's disease. The FASEB Journal 2015, 29, 589-596, 10.1096/fj.14-262048.
- Ferreira, L.S.S.; Fernandes, C.S.; Vieira, M.N.N.; de Felice, F.G; Insulin Resistance in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 830.
- Valeska Ormazabal; Soumyalekshmi Nair; Omar Elfeky; Claudio Aguayo; Carlos Salomon; Felipe Zúñiga; Association between insulin resistance and the development of cardiovascular disease. Cardiovascular Diabetology 2018, 17, 122, 10.1186/s12933-018-0762-4.
- Jean-Fred Fontaine; Adriano Barbosa-Silva; Martin H. Schaefer; Matthew Huska; Enrique M. Muro; Miguel A Andrade-Navarro; MedlineRanker: flexible ranking of biomedical literature. Nucleic Acids Research 2009, 37, W141-W146, 10.1093/nar/gkp353.
- Guanyu Wang; Raison d'être of insulin resistance: the adjustable threshold hypothesis. Journal of The Royal Society Interface 2014, 11, 20140892, 10.1098/rsif.2014.0892.
- Raid B. Nisr; Charles Affourtit; Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2014, 1837, 270-276, 10.1016/j.bbabio.2013.10.012.
- William I. Sivitz; Mark A Yorek; Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxidants & Redox Signaling 2010, 12, 537-577, 10.1089/ars.2009.2531.
- G Bucht; R Adolfsson; F Lithner; B Winblad; Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Medica Scandinavica 1983, 213, 387–392.
- Maria Niures Pimentel Dos Santos Matioli; Ricardo Nitrini; Mechanisms linking brain insulin resistance to Alzheimer's disease. Dementia & Neuropsychologia 2015, 9, 96-102, 10.1590/1980-57642015dn92000003.
- Qiu-Lan Ma; Fusheng Yang; Emily R. Rosario; Oliver J. Ubeda; Walter Beech; Dana J. Gant; Ping Ping Chen; Beverly Hudspeth; Cory Chen; Yongle Zhao; et al.Harry V. VintersSally A. FrautschyGreg M Cole Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. The Journal of Neuroscience 2009, 29, 9078-9089, 10.1523/JNEUROSCI.1071-09.2009.
- Ramon Velazquez; An Tran; Egide Ishimwe; Larry Denner; Nikhil Dave; Salvatore Oddo; Kelly T. Dineley; Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer's disease. Neurobiology of Aging 2017, 58, 1-13, 10.1016/j.neurobiolaging.2017.06.003.
- Henry H Ruiz; Tiffany Chi; Andrew C. Shin; Claudia Lindtner; Wilson Hsieh; Michelle Ehrlich; Sam Gandy; Christoph Buettner; Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer's disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimer's & Dementia 2016, 12, 851-861, 10.1016/j.jalz.2016.01.008.
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114.
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353.
- Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. Embo Mol. Med. 2015, 7, 190–210.
- Akiko Taguchi; Daisuke Tanokashira; Wataru Fukuokaya; Involvement of insulin receptor substrates in cognitive impairment and Alzheimer’s disease. Neural Regeneration Research 2019, 14, 1330-1334, 10.4103/1673-5374.253535.
- Freude, S.; Hettich, M.M.; Schumann, C.; Stohr, O.; Koch, L.; Kohler, C.; Udelhoven, M.; Leeser, U.; Muller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3315–3324.
- Akiko Taguchi; Lynn M. Wartschow; Morris F. White; Brain IRS2 Signaling Coordinates Life Span and Nutrient Homeostasis. Science 2007, 317, 369-372, 10.1126/science.1142179.
- Richard Killick; Georgie Scales; Karelle Leroy; Mirsada Causevic; Claudie Hooper; Elaine E. Irvine; Agharul I Choudhury; Laura Drinkwater; Fiona Kerr; Hind Al-Qassab; et al.John StephensonZehra YilmazK. Peter GieseJean-Pierre BrionDominic J. WithersSimon Lovestone Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochemical and Biophysical Research Communications 2009, 386, 257-262, 10.1016/j.bbrc.2009.06.032.
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1078–1089.
- Nguyen, N.H.; Pham, Q.T.; Luong, T.N.H.; Le, H.K.; Vo, V.G; Potential Antidiabetic Activity of Extracts and Isolated Compound from Adenosma bracteosum (Bonati). Biomolecules 2020, 10, 201.
- Vo Giau; Si Ying Wu; Angelo Jamerlan; Seong Soo A. An; Sangyun Kim; John Hulme; Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765, 10.3390/nu10111765.
- Ana Marta De Matos; Maria Paula Macedo; Amélia Pilar Rauter; Bridging Type 2 Diabetes and Alzheimer's Disease: Assembling the Puzzle Pieces in the Quest for the Molecules With Therapeutic and Preventive Potential. Medicinal Research Reviews 2017, 38, 261-324, 10.1002/med.21440.
- Eva Bagyinszky; Vo Van Giau; Kyuhwan Shim; Kyoungho Suk; Seong Soo A. An; Sangyun Kim; SungYun Kim; Role of inflammatory molecules in the Alzheimer's disease progression and diagnosis. Journal of the Neurological Sciences 2017, 376, 242-254, 10.1016/j.jns.2017.03.031.
- Van Giau Vo; Seong Soo A. An; John P. Hulme; Mitochondrial therapeutic interventions in Alzheimer’s disease. Journal of the Neurological Sciences 2018, 395, 62-70, 10.1016/j.jns.2018.09.033.
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11.
- Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C; Omega-3 fatty acids’ supplementation in Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2018, 21, 529–538.
- Thekkuttuparambil Ananthanarayanan Ajith; A Recent Update on the Effects of Omega-3 Fatty Acids in Alzheimer's Disease. Current Clinical Pharmacology 2018, 13, 252-260, 10.2174/1574884713666180807145648.
- Gina M. Broom; Ian C. Shaw; Julia J. Rucklidge; The ketogenic diet as a potential treatment and prevention strategy for Alzheimer's disease. Nutrition 2019, 60, 118-121, 10.1016/j.nut.2018.10.003.
- Vo Van Giau; Eva Bagyinszky; Seong Soo A. An; Sang Yun Kim; Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatric Disease and Treatment 2015, 11, 1723-1737, 10.2147/NDT.S84266.
- Stoykovich, S.; Gibas, K. APOE ε4, the door to insulin-resistant dyslipidemia and brain fog? A case study. Alzheimers Dement. (Amst) 2019, 11, 264–269.
- Na Zhao; Chia-Chen Liu; Alexandra J. Van Ingelgom; Yuka A. Martens; Cynthia Linares; Joshua A. Knight; Meghan M. Painter; Patrick M. Sullivan; Guojun Bu; Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115-129.e5, 10.1016/j.neuron.2017.09.003.
- Kristian Steen Frederiksen; Le Gjerum; Gunhild Waldemar; Steen Gregers Hasselbalch; Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. Journal of Alzheimer's Disease 2017, 61, 359-372, 10.3233/jad-170567.
- Cheng Li; Weizhen Liu; Xiaohui Li; Zijuan Zhang; Huaxin Qi; Shijin Liu; Ningning Yan; Ying Xing; Christian Hölscher; Zhiju Wang; et al. The novel GLP‐1/GIP analogue DA5‐CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain and Behavior 2020, 10, e01505, 10.1002/brb3.1505.
- Li Wang; Rui Zhang; Xiaohong Hou; Changtu Wang; Shuai Guo; Na Ning; Cong Sun; Yuan Yuan; Lin Li; Christian Hölscher; Xiao‐Hui Wang; DA-JC1 improves learning and memory by antagonizing Aβ31–35-induced circadian rhythm disorder. Molecular Brain 2019, 12, 14, 10.1186/s13041-019-0432-9.
- Yue Cao; Christian Hölscher; Meng-Ming Hu; Ting Wang; Fang Zhao; Yu Bai; Jun Zhang; Mei-Na Wu; Jinshun Qi; DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of Alzheimer's disease.. European Journal of Pharmacology 2018, 827, 215-226, 10.1016/j.ejphar.2018.03.024.
- Panagaki, T.; Gengler, S.; Holscher, C. The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1DeltaE9 Mouse Model. J. Alzheimer’s Dis. 2018, 66, 195–218.
- de Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976.
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843.
- Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al.et al Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822.
- Mark A Reger; G Stennis Watson; Pattie S Green; Laura D Baker; Brenna Cholerton; Mark A Fishel; Stephen R Plymate; Monique M Cherrier; Gerard D Schellenberg; William H Frey; et al.Suzanne Craft Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. Journal of Alzheimer's Disease 2008, 13, 323-331.
- M.A. Reger; G.S. Watson; W.H. Frey; L.D. Baker; B. Cholerton; M.L. Keeling; D.A. Belongia; M.A. Fishel; S.R. Plymate; G.D. Schellenberg; M.M. Cherrier; S. Craft; Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiology of Aging 2006, 27, 451-458, 10.1016/j.neurobiolaging.2005.03.016.
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al.et al Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448.
- Christian Benedict; Werner Kern; Bernd Schultes; Jan Born; Manfred Hallschmid; Differential Sensitivity of Men and Women to Anorexigenic and Memory-Improving Effects of Intranasal Insulin. The Journal of Clinical Endocrinology & Metabolism 2008, 93, 1339-1344, 10.1210/jc.2007-2606.
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al.et al The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non‐human primate model of Alzheimer's disease. The Journal of Pathology 2018, 245, 85-100, 10.1002/path.5056.
- Paula L. McClean; Vadivel Parthsarathy; Emilie Faivre; Christian Hölscher; The Diabetes Drug Liraglutide Prevents Degenerative Processes in a Mouse Model of Alzheimer's Disease. The Journal of Neuroscience 2011, 31, 6587-6594, 10.1523/JNEUROSCI.0529-11.2011.
- Paula L. McClean; Christian Hölscher; Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer's disease. Neuropharmacology 2014, 76, 57-67, 10.1016/j.neuropharm.2013.08.005.
- TracyAnn Perry; Norman J. Haughey; Mark P. Mattson; Josephine M. Egan; Nigel H. Greig; Protection and Reversal of Excitotoxic Neuronal Damage by Glucagon-Like Peptide-1 and Exendin-4. Journal of Pharmacology and Experimental Therapeutics 2002, 302, 881-888, 10.1124/jpet.102.037481.
- Luis Escribano; Ana-María Simón; Esther Gimeno; Mar Cuadrado-Tejedo; Rakel López De Maturana; Ana García-Osta; Ana Ricobaraza; Alberto Pérez-Mediavilla; Joaquín Del Río; Diana Frechilla; Rosiglitazone Rescues Memory Impairment in Alzheimer's Transgenic Mice: Mechanisms Involving a Reduced Amyloid and Tau Pathology. Neuropsychopharmacology 2010, 35, 1593-1604, 10.1038/npp.2010.32.