Osteoarthritis (OA) is a chronic degenerative joint disease where the main characteristics include cartilage degeneration and synovial membrane inflammation.
- osteoarthritis
- articular cartilage
1. Introduction
Osteoarthritis (OA) is a common joint disease where the primary risk factors include traumatic joint injury or the mechanical disruption of joint tissues due to accumulated external forces. The etiology is multifactorial, and both environmental and genetic factors are thought to contribute to triggering the pathogenesis and progression of OA [1]. Critical hallmarks of OA include cartilage degeneration, osteophyte formation, and fibrosis in the joint tissue [2]. When left untreated, the defected joint eventually requires total joint replacement.
Tissue fibrosis occurs in various tissues, including the liver, kidney, heart, and others, and it eventually leads to organ failure. Fibrosis also occurs in the articular joint during the progression of OA and plays a critical role in OA pathogenesis and progression as well as cartilage destruction [3]. Joint fibrosis is characterized by the excessive accumulation of connective tissue, which eventually contributes to joint stiffness that results in intense pain during joint movement [4]. While the articular cartilage is unable to repair itself, affected chondrocytes often undergo de-differentiation and convert into fibrotic chondrocytes. Then, fibrotic chondrocytes secrete proteins that are similar to fibrocartilage, which is stiffer and mechanically inferior compared to the original hyaline cartilage [5,6][5][6]. Then, the cartilage defect recovers with fibrocartilage-like tissue that lacks its original function and even worsens the symptoms of OA [7]. This process also occurs in the cells (e.g., primary chondrocytes, mesenchymal stem cells) that are applied for OA therapy [8]. Synovitis caused by hyperplasia in the synovial membrane is also thought to be related to fibrosis [9].
2. Fibrosis and OA
Fibrosis is usually a wound healing process [3]. The classic wound healing process results in cell proliferation, differentiation, ECM production, and remodeling. However, an abnormal wound healing process can lead to the excessive secretion and deposition of ECM proteins in the tissue, followed by excessive fibrosis, which eventually results in scar formation, inflammation, and even damage in the tissue [40][10].
Fibrosis that occurs during OA induces joint stiffness and pain. As mentioned earlier, fibrosis is found in two events related to OA: (1) the generation of fibrotic cartilage during cartilage repair or OA progression (Figure 1A) and (2) synovial fibrosis during the onset and progression of OA (Figure 1B).
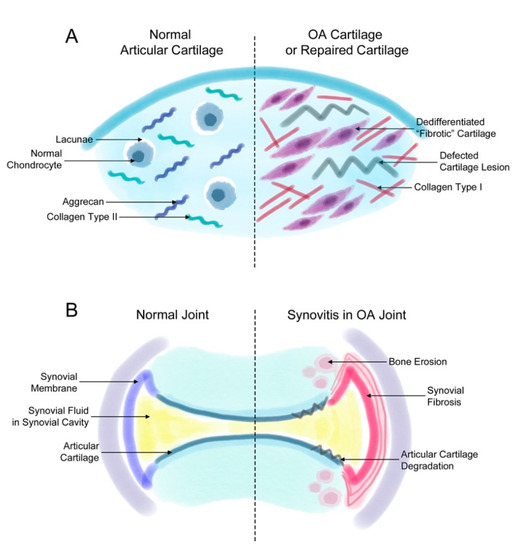
Figure 1. Fibrosis in synovial and cartilage tissue. (A) The normal joint consists of a smooth layer of articular cartilage and a smooth layer of synovial membrane on the side that maintains the synovial fluid in the synovial cavity. In the joint of a patient with osteoarthritis (OA), the increased proliferation of synovial cells induces synovial fibrosis that results in joint swelling, stiffness, and pain. The increasing levels of synovium eventually affect the cartilage and bone tissue, inducing further degradation of the cartilage tissue and bone erosion. (B) The normal articular cartilage has a smooth extracellular matrix (ECM) that is mostly composed of aggrecan and collagen type II. Normally, chondrocytes remain in small spaces called lacunae. However, chondrocytes in a defected cartilage lesion undergo abnormal proliferation that leads to their de-differentiation into fibroblast-like fibrotic chondrocytes. Then, these chondrocytes secrete ECM proteins such as collagen type I instead of aggrecan or collagen type II. These changes lead to a stiffer type of cartilage and eventually completely change the characteristics of the articular cartilage.
3. Fibrosis-Related Markers in OA
Two main factors that are critical for fibrotic disease, namely TGFβ and connective tissue growth factor (CTGF), are also elevated in OA and are the main candidates for the development of joint fibrosis. The overexpression of CTGF in the joints of mice induced synovial fibrosis [63][11]. CTGF expression can be induced by TGFβ; however, it can also function independently [2,3,71][2][3][12]. Scharstuhl et al. showed that TGFβ plays an important role in synovial thickening by fibrosis in experimental OA, and also that blocking this pathway prevented this process [72][13]. TGFβ promotes the formation of ECM by inducing collagen and fibronectin synthesis [73][14]. TGFβ also plays a critical role in fibrosis in various organs and is known to control cell proliferation, differentiation, immunity, and wound healing [2]. While TGFβ plays a critical role in cartilage fibrosis in patients with OA, blocking this molecule is not a treatment option since it is also a critical factor necessary for chondrogenesis. High levels of TGFβ exist in healthy cartilage, while low levels are found in OA cartilage [74][15]. Adequate levels of TGFβ even had protective effects on cartilage in animal models of arthritis; however, an excessive amount of this growth factor had adverse effects [75][16]. It is also reported that high concentrations of TGFβ exist in OA synovial fluids, which is produced by synoviocytes [76][17]. The administration of 20 ng of TGFβ was enough to increase the number of synovial lining cells by inducing fibroblast proliferation, along with collagen deposition [77][18]. The delivery of TGFβ by injection or transfection also resulted in increased synovial hyperplasia and osteophyte formation [78][19]. High levels of TGFβ activate the SMAD1-5-8 pathway instead of the SMAD2-3 pathway. The activation of the SMAD1-5-8 pathway upregulates genes related to fibrogenic differentiation and hypertrophy, and eventually induces synovial fibrosis and osteophyte formation [79][20]. However, it is reported that the difference between the two major growth factors is that fibrosis induced by TGFβ is relatively persistent, while CTGF-induced fibrosis is relatively temporary (about 28 days) [2]. Moreover, the viral delivery of TGFβ increased COL1A1 expression, but CTGF overexpression did not [80][21].
Collagen type I is also a well-known marker for fibrosis and has been detected in various fibrosis-related diseases, including OA [81,82,83][22][23][24]. Collagen type I and alpha-smooth muscle actin (α-SMA) are key markers of both fibrocartilage formation and synovial fibrosis [84][25]. Remodeled cartilage in the defects of osteoarthritic cartilage show fibrotic characteristics such as increased levels of collagen type I and α-SMA [85,86,87][26][27][28]. Collagen type I is a major marker of unfavorable fibrocartilage and chondrocyte de-differentiation [8,88][8][29]. As mentioned earlier, in vitro de-differentiated chondrocytes express the OA-related de-differentiation marker collagen type I. Articular chondrocytes that undergo longer expansion times with multiple passages in monolayer culture end up in chondrocyte de-differentiation [89][30]. During the de-differentiation process, chondrocytes go through morphological and phenotypical changes and become fibroblastic, eventually producing collagen type I [88][29]. The compositional changes in the ECM also affect the chondrogenicity of MSCs [43][31]. MSCs cultured in collagen type II hydrogels showed higher expression of chondrogenic markers, which might indicate chondrocyte de-differentiation caused by collagen type I [90][32]. During OA progression, residential chondrocytes producing collagen type I might affect the subsequent chondrogenesis of MSCs [91][33]. Collagen type I is the major marker for fibrotic de-differentiated hyaline cartilage, but collagen type III is also thought to be related to the de-differentiation process, which suggests that it could also be a marker for the fibroblast-like phenotype. TGFβ1 markedly increased the SMA content in chondrocytes [92][34]. The secretion of α-SMA by OA chondrocytes has been verified and was observed to significantly increase in OA cartilage, along with collagen type III [51,84][25][35].
The de-differentiated chondrocytes and fibrocartilage formed in the articular cartilage lesion produce pro-inflammatory mediators that further induce cartilage degeneration and synovial inflammation. IL-1β has been identified as the most prevalent cytokine in OA, which is involved in cell proliferation, differentiation, and apoptosis [93][36]. IL-1β also interferes with the production of hyaline cartilage structural proteins, collagen type II, and aggrecan by altering chondrocyte characteristics.
Proteases are actively involved in the pathogenesis of OA. Proteases, including matrix metallopeptidases (MMPs), disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs), mainly degrade the main hyaline cartilage ECM proteins aggrecan and collagen type II [94][37]. MMPs are a zinc-dependent enzyme family that degrades ECM proteins in the articular cartilage. Various types of MMPs exist and can be categorized into several groups: collagenases, gelatinases, stromelysins, metalloelastases, and more. Of all the MMPs, MMP13 is considered to be the most important agent for collagen degradation that leads to OA, as it degrades collagen type II [95,96][38][39]. Some MMPs have also been proposed to regulate ADAMTS activity [97][40]. ADAMTSs eventually circulate in a vital cycle between the degenerated chondrocyte and other cells near it, such as synoviocytes in the synovial membrane. These proteases are also reported to be related to fibrosis in several diseases including idiopathic pulmonary fibrosis, liver fibrosis, and others [98,99][41][42]. Therefore, they are also thought to be related to joint fibrosis; however, there is currently little information on the direct relationship between the two factors.
Other markers such as procollagen lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2), and tissue inhibitor of metalloproteinase 1 (TIMP1) are reported to increase in OA-related fibrosis and are also considered fibrotic markers [3,100][3][43]. PLOD2 is an enzyme related to collagen crosslinking that induces pyrodinoline crosslink formation [3]. Pyrodinoline crosslinks make collagen fibers resistant to collagen degradation, which makes them more rigid and results in collagen accumulation in fibrotic tissues [101][44]. Collagen accumulation is induced by reduced levels of collagen degradation, inducing increased pyridinoline crosslinks per collagen triple helix [101][44]. Remst et al. confirmed the presence of increased levels of PLOD2 and an increased pyridinoline crosslink/collagen ratio in OA synovium, and this expression increased along with the severity of OA. While the presence of fibrosis in OA patients was unknown, it is expected that PLOD2 may have higher expression in the subpopulation of OA patients with fibrosis. Ueki et al. confirmed that the knockout of PLOD2 inhibited the tumorigenesis role of integrin β1 in tumor cells [102][45]. While it is still controversial, integrins are reported to be closely related to the pathogenesis of OA, and the confirmation of this process in OA might suggest a new candidate for OA treatment [103,104][46][47]. TIMP1 is an inhibitor of matrix metallopeptidases (MMPs), which are well known for their role in OA. The elevation of TIMP1 expression was confirmed in various fibrotic diseases, including synovial fibrosis in OA [105][48]. TIMP1 elevation in fibrotic tissue is induced by elevated levels of TGFβ, and TIMP1 induces fibrosis via the downregulation of MMPs [106][49]. The relationship between hypoxia and synovial fibrosis has been also shown by observing the expression of HIF-1α; the expression of TGFβ, COL1A1, PLOD2, and TIMP1 was downregulated by the inhibition of HIF-1α in rat models of OA [107][50].
References
- Liu, H.; Yang, L.; Yu, F.F.; Wang, S.; Wu, C.; Qu, C.; Lammi, M.J.; Guo, X. The potential of induced pluripotent stem cells as a tool to study skeletal dysplasias and cartilage-related pathologic conditions. Osteoarthr. Cartil. 2017, 25, 616–624.
- Remst, D.F.; Blaney Davidson, E.N.; Vitters, E.L.; Blom, A.B.; Stoop, R.; Snabel, J.M.; Bank, R.A.; van den Berg, W.B.; van der Kraan, P.M. Osteoarthritis-related fibrosis is associated with both elevated pyridinoline cross-link formation and lysyl hydroxylase 2b expression. Osteoarthr. Cartil. 2013, 21, 157–164.
- Remst, D.F.; Blaney Davidson, E.N.; van der Kraan, P.M. Unravelling osteoarthritis-related synovial fibrosis: A step closer to solving joint stiffness. Rheumatology 2015, 54, 1954–1963.
- Hill, C.L.; Hunter, D.J.; Niu, J.; Clancy, M.; Guermazi, A.; Genant, H.; Gale, D.; Grainger, A.; Conaghan, P.; Felson, D.T. Synovitis detected on magnetic resonance imaging and its relation to pain and cartilage loss in knee osteoarthritis. Ann. Rheum. Dis. 2007, 66, 1599–1603.
- Phull, A.R.; Eo, S.H.; Abbas, Q.; Ahmed, M.; Kim, S.J. Applications of Chondrocyte-Based Cartilage Engineering: An Overview. Biomed. Res. Int. 2016, 2016, 1879837.
- Dell’Accio, F.; De Bari, C.; Luyten, F.P. Microenvironment and phenotypic stability specify tissue formation by human articular cartilage-derived cells in vivo. Exp. Cell Res. 2003, 287, 16–27.
- Karuppal, R. Current concepts in the articular cartilage repair and regeneration. J. Orthop. 2017, 14, A1–A3.
- Armiento, A.R.; Alini, M.; Stoddart, M.J. Articular fibrocartilage—Why does hyaline cartilage fail to repair? Adv. Drug Deliv. Rev. 2019, 146, 289–305.
- Wenham, C.Y.; Conaghan, P.G. The role of synovitis in osteoarthritis. Adv. Musculoskelet. Dis. 2010, 2, 349–359.
- Chan, D.D.; Li, J.; Luo, W.; Predescu, D.N.; Cole, B.J.; Plaas, A. Pirfenidone reduces subchondral bone loss and fibrosis after murine knee cartilage injury. J. Orthop. Res. 2018, 36, 365–376.
- Blaney Davidson, E.N.; Vitters, E.L.; Mooren, F.M.; Oliver, N.; Berg, W.B.; van der Kraan, P.M. Connective tissue growth factor/CCN2 overexpression in mouse synovial lining results in transient fibrosis and cartilage damage. Arthritis Rheum. 2006, 54, 1653–1661.
- Blom, I.E.; Goldschmeding, R.; Leask, A. Gene regulation of connective tissue growth factor: New targets for antifibrotic therapy? Matrix Biol. 2002, 21, 473–482.
- Scharstuhl, A.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Reduction of osteophyte formation and synovial thickening by adenoviral overexpression of transforming growth factor beta/bone morphogenetic protein inhibitors during experimental osteoarthritis. Arthritis Rheum. 2003, 48, 3442–3451.
- Xue, M.; Gong, S.; Dai, J.; Chen, G.; Hu, J. The Treatment of Fibrosis of Joint Synovium and Frozen Shoulder by Smad4 Gene Silencing in Rats. PLoS ONE 2016, 11, e0158093.
- Verdier, M.P.; Seite, S.; Guntzer, K.; Pujol, J.P.; Boumediene, K. Immunohistochemical analysis of transforming growth factor beta isoforms and their receptors in human cartilage from normal and osteoarthritic femoral heads. Rheumatol. Int. 2005, 25, 118–124.
- Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; van den Berg, W.B. Stimulation of articular cartilage repair in established arthritis by local administration of transforming growth factor-beta into murine knee joints. Lab. Investig. 1998, 78, 133–142.
- Ruiz, M.; Maumus, M.; Fonteneau, G.; Pers, Y.M.; Ferreira, R.; Dagneaux, L.; Delfour, C.; Houard, X.; Berenbaum, F.; Rannou, F.; et al. TGFbetai is involved in the chondrogenic differentiation of mesenchymal stem cells and is dysregulated in osteoarthritis. Osteoarthr. Cartil. 2019, 27, 493–503.
- Allen, J.B.; Manthey, C.L.; Hand, A.R.; Ohura, K.; Ellingsworth, L.; Wahl, S.M. Rapid onset synovial inflammation and hyperplasia induced by transforming growth factor beta. J. Exp. Med. 1990, 171, 231–247.
- van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthr. Cartil. 2000, 8, 25–33.
- van der Kraan, P.M. The changing role of TGFbeta in healthy, ageing and osteoarthritic joints. Nat. Rev. Rheumatol. 2017, 13, 155–163.
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Haberberger, T.; Kelly, M.; Robertson, J.; Gauldie, J. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 770–778.
- Trojanowska, M.; LeRoy, E.C.; Eckes, B.; Krieg, T. Pathogenesis of fibrosis: Type 1 collagen and the skin. J. Mol. Med. 1998, 76, 266–274.
- Hansen, N.U.; Karsdal, M.A.; Brockbank, S.; Cruwys, S.; Ronnow, S.; Leeming, D.J. Tissue turnover of collagen type I, III and elastin is elevated in the PCLS model of IPF and can be restored back to vehicle levels using a phosphodiesterase inhibitor. Respir. Res. 2016, 17, 76.
- Wu, L.; Petrigliano, F.A.; Ba, K.; Lee, S.; Bogdanov, J.; McAllister, D.R.; Adams, J.S.; Rosenthal, A.K.; Van Handel, B.; Crooks, G.M.; et al. Lysophosphatidic acid mediates fibrosis in injured joints by regulating collagen type I biosynthesis. Osteoarthr. Cartil. 2015, 23, 308–318.
- Liu, N.; Fu, D.; Yang, J.; Liu, P.; Song, X.; Wang, X.; Li, R.; Fu, Z.; Chen, J.; Gong, X.; et al. Asiatic acid attenuates hypertrophic and fibrotic differentiation of articular chondrocytes via AMPK/PI3K/AKT signaling pathway. Arthritis Res. Ther. 2020, 22, 112.
- Adam, M.; Deyl, Z. Altered expression of collagen phenotype in osteoarthrosis. Clin. Chim. Acta 1983, 133, 25–32.
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65.
- Gay, S.; Muller, P.K.; Lemmen, C.; Remberger, K.; Matzen, K.; Kuhn, K. Immunohistological study on collagen in cartilage-bone metamorphosis and degenerative osteoarthrosis. Klin. Wochenschr. 1976, 54, 969–976.
- Giannoni, P.; Cancedda, R. Articular chondrocyte culturing for cell-based cartilage repair: Needs and perspectives. Cells Tissues Organs 2006, 184, 1–15.
- Tallheden, T.; Bengtsson, C.; Brantsing, C.; Sjogren-Jansson, E.; Carlsson, L.; Peterson, L.; Brittberg, M.; Lindahl, A. Proliferation and differentiation potential of chondrocytes from osteoarthritic patients. Arthritis Res. Ther. 2005, 7, R560–R568.
- Maldonado, M.; Nam, J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. BioMed. Res. Int. 2013, 2013, 284873.
- Bosnakovski, D.; Mizuno, M.; Kim, G.; Takagi, S.; Okumura, M.; Fujinaga, T. Chondrogenic differentiation of bovine bone marrow mesenchymal stem cells (MSCs) in different hydrogels: Influence of collagen type II extracellular matrix on MSC chondrogenesis. Biotechnol. Bioeng. 2006, 93, 1152–1163.
- Lu, Z.; Doulabi, B.Z.; Huang, C.; Bank, R.A.; Helder, M.N. Collagen type II enhances chondrogenesis in adipose tissue-derived stem cells by affecting cell shape. Tissue Eng. Part A 2010, 16, 81–90.
- Zaleskas, J.M.; Kinner, B.; Freyman, T.M.; Yannas, I.V.; Gibson, L.J.; Spector, M. Growth factor regulation of smooth muscle actin expression and contraction of human articular chondrocytes and meniscal cells in a collagen-GAG matrix. Exp. Cell Res. 2001, 270, 21–31.
- Deroyer, C.; Charlier, E.; Neuville, S.; Malaise, O.; Gillet, P.; Kurth, W.; Chariot, A.; Malaise, M.; de Seny, D. CEMIP (KIAA1199) induces a fibrosis-like process in osteoarthritic chondrocytes. Cell Death Dis. 2019, 10, 103.
- Chow, Y.Y.; Chin, K.Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 8293921.
- Murphy, G.; Lee, M.H. What are the roles of metalloproteinases in cartilage and bone damage? Ann. Rheum. Dis. 2005, 64 (Suppl. 4), iv44–iv47.
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786.
- Griffin, F.M.; Math, K.; Scuderi, G.R.; Insall, J.N.; Poilvache, P.L. Anatomy of the epicondyles of the distal femur: MRI analysis of normal knees. J. Arthroplast. 2000, 15, 354–359.
- Patwari, P.; Gao, G.; Lee, J.H.; Grodzinsky, A.J.; Sandy, J.D. Analysis of ADAMTS4 and MT4-MMP indicates that both are involved in aggrecanolysis in interleukin-1-treated bovine cartilage. Osteoarthr. Cartil. 2005, 13, 269–277.
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727.
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 53, 585–600.
- Zhang, L.; Xing, R.; Huang, Z.; Zhang, N.; Zhang, L.; Li, X.; Wang, P. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediat. Inflamm. 2019, 2019, 2165918.
- van der Slot, A.J.; Zuurmond, A.M.; van den Bogaerdt, A.J.; Ulrich, M.M.; Middelkoop, E.; Boers, W.; Karel Ronday, H.; DeGroot, J.; Huizinga, T.W.; Bank, R.A. Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004, 23, 251–257.
- Ueki, Y.; Saito, K.; Iioka, H.; Sakamoto, I.; Kanda, Y.; Sakaguchi, M.; Horii, A.; Kondo, E. PLOD2 Is Essential to Functional Activation of Integrin beta1 for Invasion/Metastasis in Head and Neck Squamous Cell Carcinomas. iScience 2020, 23, 100850.
- Loeser, R.F. Integrins and chondrocyte-matrix interactions in articular cartilage. Matrix Biol. 2014, 39, 11–16.
- Zhang, L.Q.; Zhao, G.Z.; Xu, X.Y.; Fang, J.; Chen, J.M.; Li, J.W.; Gao, X.J.; Hao, L.J.; Chen, Y.Z. Integrin-beta1 regulates chondrocyte proliferation and apoptosis through the upregulation of GIT1 expression. Int. J. Mol. Med. 2015, 35, 1074–1080.
- Remst, D.F.; Blom, A.B.; Vitters, E.L.; Bank, R.A.; van den Berg, W.B.; Blaney Davidson, E.N.; van der Kraan, P.M. Gene expression analysis of murine and human osteoarthritis synovium reveals elevation of transforming growth factor beta-responsive genes in osteoarthritis-related fibrosis. Arthritis Rheumatol. 2014, 66, 647–656.
- Yoshiji, H.; Kuriyama, S.; Miyamoto, Y.; Thorgeirsson, U.P.; Gomez, D.E.; Kawata, M.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Tsujinoue, H.; et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology 2000, 32, 1248–1254.
- Zhang, L.; Zhang, L.; Huang, Z.; Xing, R.; Li, X.; Yin, S.; Mao, J.; Zhang, N.; Mei, W.; Ding, L.; et al. Increased HIF-1alpha in Knee Osteoarthritis Aggravate Synovial Fibrosis via Fibroblast-Like Synoviocyte Pyroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 6326517.