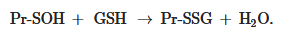1. Introduction
Redox-dependent processes largely determine cell viability, participating in the regulation of division, bioenergetics, and programmed death. The cellular redox status is characterized by low-molecular-weight indicators (GSH, NADH). The change in their oxidized/reduced form ratio occurs as a reaction to changes in reactive oxygen and nitrogen species (RONS) levels and, so, they can play the role of a trigger in the redox-dependent regulation of cellular processes. Undoubtedly, such an important trigger role is played by glutathione (γ-glutamyl-
l
-cysteinylglycine, GSH), a water-soluble tripeptide consisting of the amino acids
l
-glutamate,
l-cysteine, and glycine, which is widely present in both eukaryotes and prokaryotes[1][2][3]. GSH is less susceptible to oxidation than Cys, which makes it the most suitable for maintaining intracellular redox status[1]. The presence of a γ-peptide bond at the Glu residue protects GSH from the action of peptidases, while the SH group at the Cys residue makes GSH a good electron donor, allowing it to participate in reactions with strong electrophiles.
-cysteine, and glycine, which is widely present in both eukaryotes and prokaryotes [1,2,3]. GSH is less susceptible to oxidation than Cys, which makes it the most suitable for maintaining intracellular redox status [1]. The presence of a γ-peptide bond at the Glu residue protects GSH from the action of peptidases, while the SH group at the Cys residue makes GSH a good electron donor, allowing it to participate in reactions with strong electrophiles.
Normally, the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG), GSH/GSSG—which characterizes the cellular redox status—is 100/1 in the cytoplasm, 10/1 in mitochondria, and 3/1 to 1 in the endoplasmic reticulum[4]. This ratio varies depending on the physiological state of cells, such as proliferation, differentiation, or apoptosis, and the consequences of its disturbance are significant changes in cellular signaling reactions. This role of GSH/GSSG is largely due to its regulatory effect on the functional activities of protein thiols[5][6][7].
Normally, the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG), GSH/GSSG—which characterizes the cellular redox status—is 100/1 in the cytoplasm, 10/1 in mitochondria, and 3/1 to 1 in the endoplasmic reticulum [4]. This ratio varies depending on the physiological state of cells, such as proliferation, differentiation, or apoptosis, and the consequences of its disturbance are significant changes in cellular signaling reactions. This role of GSH/GSSG is largely due to its regulatory effect on the functional activities of protein thiols [5,6,7].
Although Cys residues in mammalian proteins do not exceed 3%[6], they are highly sensitive to oxidative modification, which significantly affects the functioning of proteins, as thiol groups play a significant role in the formation of protein tertiary and quaternary structures and enzyme active sites. The pKa value of most SH groups of cellular proteins is more than 8.0, which keeps thiol groups predominantly protonated at physiological pH values (pH 7.0–7.4)[8][9]. However, in proteins, in the immediate vicinity of the basic amino acid residues (histidine, lysine, and arginine), the pKa of the SH groups decreases (usually to 5.0–7.0) and these thiols dissociate at physiological pH. The resulting thiolate anions (Pr-S
Although Cys residues in mammalian proteins do not exceed 3% [6], they are highly sensitive to oxidative modification, which significantly affects the functioning of proteins, as thiol groups play a significant role in the formation of protein tertiary and quaternary structures and enzyme active sites. The pKa value of most SH groups of cellular proteins is more than 8.0, which keeps thiol groups predominantly protonated at physiological pH values (pH 7.0–7.4) [8,9]. However, in proteins, in the immediate vicinity of the basic amino acid residues (histidine, lysine, and arginine), the pKa of the SH groups decreases (usually to 5.0–7.0) and these thiols dissociate at physiological pH. The resulting thiolate anions (Pr-S −) are effective nucleophiles and have high activity with respect to electrophilic targets[10][11][12]. The reactivity of SH groups and the functional activity of proteins are largely regulated by S-glutathionylation and S-nitrosylation[6][13][14][15][16].
) are effective nucleophiles and have high activity with respect to electrophilic targets [10,11,12]. The reactivity of SH groups and the functional activity of proteins are largely regulated by S-glutathionylation and S-nitrosylation [6,13,14,15,16].
Under S-glutathionylation, GSH can bind to the cysteinyl residues of proteins through the creation of reversible disulfide bonds, depending on the cysteine position and redox potential [12][13]. This post-translational modification to the protein can lead to enhanced or suppressed activity, may prevent protein degradation by proteolysis or sulfhydryl overoxidation, and plays an important role in cellular signaling. At present, the dual role of S-glutathionylation in maintaining cellular homeostasis and participating in various pathological processes may be indicated[14][17].
Under S-glutathionylation, GSH can bind to the cysteinyl residues of proteins through the creation of reversible disulfide bonds, depending on the cysteine position and redox potential [12,13]. This post-translational modification to the protein can lead to enhanced or suppressed activity, may prevent protein degradation by proteolysis or sulfhydryl overoxidation, and plays an important role in cellular signaling. At present, the dual role of S-glutathionylation in maintaining cellular homeostasis and participating in various pathological processes may be indicated [14,17].
Under S-nitrosylation, NO is covalently attached to the SH group of a cysteine residue and, as a consequence, can cause alterations in the cellular function of a variety of proteins[18][19]. S-nitrosoglutathione, formed as the result of GSH S-nitrosylation, serves as a NO reservoir and can transfer NO groups to new cysteine residues in transnitrosylation reactions[20][21].
Under S-nitrosylation, NO is covalently attached to the SH group of a cysteine residue and, as a consequence, can cause alterations in the cellular function of a variety of proteins [18,19]. S-nitrosoglutathione, formed as the result of GSH S-nitrosylation, serves as a NO reservoir and can transfer NO groups to new cysteine residues in transnitrosylation reactions [20,21].
The GSH/GSSG ratio can be considered a key redox sensor, which determines the redox-dependent alteration of the protein functional activity through such significant post-translational modifications as S-glutathionylation and S-nitrosylation. The modulation of the activity of these reactions in response to a change in the GSH/GSSG ratio, as a result of an increase or decrease in RONS levels, provides a significant contribution to cell functional adaptation to redox changes of the environment [22][23][24][25].
The GSH/GSSG ratio can be considered a key redox sensor, which determines the redox-dependent alteration of the protein functional activity through such significant post-translational modifications as S-glutathionylation and S-nitrosylation. The modulation of the activity of these reactions in response to a change in the GSH/GSSG ratio, as a result of an increase or decrease in RONS levels, provides a significant contribution to cell functional adaptation to redox changes of the environment [22,23,24,25].
In this review, we analyze data concerning the roles of GSH and GSH/GSSG in the redox modulation of S-glutathionylation and S-nitrosylation and their relationship for the maintenance of cell viability.
2. Glutathione and Protein S-Glutathionylation
A widespread form of cysteine modification is S-glutathionylation—the reversible formation of protein mixed disulfides with GSH (Pr-SSG)—which occurs in the cell under physiological conditions and oxidative stress, both spontaneously and enzymatically. The S-glutathionylation of proteins suggests the possible involvement of this post-translational modification in cellular signaling and the redox regulation of protein functions[6][13][24]. In addition to the potential regulatory role, S-glutathionylation can serve as a means of GSH storage, as well as protection from the irreversible oxidation of protein thiol groups under stress conditions[26], often due to a temporary loss of primary protein activity as, if the modified sulfhydryl group of a protein is functionally critical, S-glutathionylation can render the protein inactive or alter its activity, ultimately disrupting cellular functions[27]. In addition, this reaction can affect a change in conformation and/or charge, which can modify the function of the protein, as the attachment of GSH introduces an additional negative charge at the expense of the glutamic acid residue.
A widespread form of cysteine modification is S-glutathionylation—the reversible formation of protein mixed disulfides with GSH (Pr-SSG)—which occurs in the cell under physiological conditions and oxidative stress, both spontaneously and enzymatically. The S-glutathionylation of proteins suggests the possible involvement of this post-translational modification in cellular signaling and the redox regulation of protein functions [6,13,24]. In addition to the potential regulatory role, S-glutathionylation can serve as a means of GSH storage, as well as protection from the irreversible oxidation of protein thiol groups under stress conditions [26], often due to a temporary loss of primary protein activity as, if the modified sulfhydryl group of a protein is functionally critical, S-glutathionylation can render the protein inactive or alter its activity, ultimately disrupting cellular functions [27]. In addition, this reaction can affect a change in conformation and/or charge, which can modify the function of the protein, as the attachment of GSH introduces an additional negative charge at the expense of the glutamic acid residue.
Non-enzymatic S-glutathionylation reactions can occur during thiol-disulfide exchange, through the participation of protein thiol (Pr-SH) and oxidized glutathione GSSG:
Non-enzymatic S-glutathionylation reactions can occur during thiol-disulfide exchange, through the participation of protein thiol (Pr-SH) and oxidized glutathione GSSG:
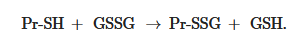 GSS
GSS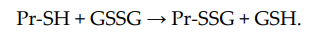
The equilibrium constant of the reaction K
mix is expressed by the ratio [Pr-SSG]·[GSH]/[Pr-SH]·[GSSG], where the extent of S-glutathionylated proteins ([Pr-SSG]/[Pr-SH]) strongly depends on the local ratio [GSH]/[GSSG][28][29]. The S-glutathionylation of most proteins with typical redox potential (K
is expressed by the ratio [Pr-SSG]·[GSH]/[Pr-SH]·[GSSG], where the extent of S-glutathionylated proteins ([Pr-SSG]/[Pr-SH]) strongly depends on the local ratio [GSH]/[GSSG] [28,29]. The S-glutathionylation of most proteins with typical redox potential (K mix~1) by about 50% can occur when this ratio drops very dramatically (i.e., from 100:1 to 1:1). These extreme conditions are rare in vivo. Therefore, for most proteins, the spontaneous formation of Pr-SSG—as a result of the exchange of Pr-SH and GSSG—is not common and, as a rule, takes place under pathological conditions[30]. In addition, a variant of thiol-disulfide exchange between Pr-SH and a protein which is already S-glutathionylated (Pr′-SSG) is possible:
~1) by about 50% can occur when this ratio drops very dramatically (i.e., from 100:1 to 1:1). These extreme conditions are rare in vivo. Therefore, for most proteins, the spontaneous formation of Pr-SSG—as a result of the exchange of Pr-SH and GSSG—is not common and, as a rule, takes place under pathological conditions [30]. In addition, a variant of thiol-disulfide exchange between Pr-SH and a protein which is already S-glutathionylated (Pr′-SSG) is possible:
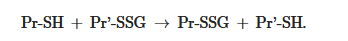
SHSH.SSG → Pr-
S-glutathionylation can occur when Pr-SH or GSH reacts with an oxidized derivative of the protein cysteine residue; for example, sulfenic acid (-SOH), thiyl radical (-S
•
), or S-nitrosyl (-SNO) group. Thus, when Pr-SH is oxidized with, for example, H
2
O
2
, sulfenic acid (Pr-SOH) is formed and then quickly reacts with GSH to form Pr-SSG:
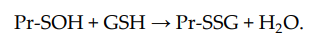
Sulfenic acid is unstable and can undergo further oxidation to sulfinic acid (Pr-SO
Sulfenic acid is unstable and can undergo further oxidation to sulfinic acid (Pr-SO
2
H) and eventually to sulfonic acid (Pr-SO
3H), the formation of which, as a rule, leads to the irreversible deactivation of the protein. Thus, the S-glutathionylation of sulfenic acid can prevent the oxidation of protein cysteine residues[6][25]. Under physiological conditions, the intracellular level of H
H), the formation of which, as a rule, leads to the irreversible deactivation of the protein. Thus, the S-glutathionylation of sulfenic acid can prevent the oxidation of protein cysteine residues [6,25]. Under physiological conditions, the intracellular level of H 2
O
2
is in the sub-micromolar range (10
−9
–10
−7
M) [31]. Therefore, in vivo spontaneous S-glutathionylation proceeds rather slowly by this mechanism.
The formation of S-glutathionylated protein is also possible, due to its interaction with GSSG in the form of sulfenic acid (GSOH):
Thus, it is obvious that the S-glutathionylation of proteins can occur spontaneously; however, the rate and extent of this process increases with the participation of enzymes, among which glutathione transferase isoform P1-1 (GSTP1-1) plays the greatest role[32][33]. GSTP1-1 has been shown to facilitate S-glutathionylation for a number of proteins, including peroxiredoxin 6 (Prx6)[34][35], aldose reductase[36], actin[37], histone H3[38], 5’AMP-activated protein kinase (AMPK)[32], estrogen receptor α[39], heat shock protein BiP, protein disulfide isomerase (PDI), calnexin, calreticulin, and sarcoplasmic reticulum Ca
Thus, it is obvious that the S-glutathionylation of proteins can occur spontaneously; however, the rate and extent of this process increases with the participation of enzymes, among which glutathione transferase isoform P1-1 (GSTP1-1) plays the greatest role [32,33]. GSTP1-1 has been shown to facilitate S-glutathionylation for a number of proteins, including peroxiredoxin 6 (Prx6) [34,35], aldose reductase [36], actin [37], histone H3 [38], 5’AMP-activated protein kinase (AMPK) [32], estrogen receptor α [39], heat shock protein BiP, protein disulfide isomerase (PDI), calnexin, calreticulin, and sarcoplasmic reticulum Ca 2+-ATPase (SERCA)[33].
During the reaction, GSTP1-1 binds GSH in the active center and decreases the pKa of the GSH cysteine residue from 9.2 to 6.3[40], deprotonates it with the participation of Tyr7, forming a thiolate anion (GS
During the reaction, GSTP1-1 binds GSH in the active center and decreases the pKa of the GSH cysteine residue from 9.2 to 6.3 [40], deprotonates it with the participation of Tyr7, forming a thiolate anion (GS −), which is transferred to the cysteine residue in the substrate. In cells with the Tyr7 GSTP1-1 mutation, a decrease in the total content of S-glutathionylated proteins has been observed upon treatment with GSSG, the mimetic of oxidized glutathione NOV-002 and diazeniumdiolate-based NO-donor prodrug PABA/NO[33]. An example is the S-glutathionylation of Prx6 from the 1-Cys groups of peroxiredoxins. As a result of human Prx6 peroxidase activity, the Cys47 in the active center is oxidized to sulfenic acid; this deprives it of activity; as for the reduction of which, a second thiol is required to form a mixed disulfide, then a sulfhydryl group. However, the availability of the sulfenic group is low, due to the peculiarities of the globular structure of Prx6. Prx6 activation occurs during the formation of a heterodimer with GSTP1-1, which promotes the S-glutathionylation of Cys47 Prx6. The conformational changes of the heterodimer occur, providing the formation of a disulfide bond between Cys47 GSTP1-1 and Cys47 Prx6, followed by the reduction of disulfide with the participation of GSH and the regeneration of Cys47 Prx6[34].
), which is transferred to the cysteine residue in the substrate. In cells with the Tyr7 GSTP1-1 mutation, a decrease in the total content of S-glutathionylated proteins has been observed upon treatment with GSSG, the mimetic of oxidized glutathione NOV-002 and diazeniumdiolate-based NO-donor prodrug PABA/NO [36]. An example is the S-glutathionylation of Prx6 from the 1-Cys groups of peroxiredoxins. As a result of human Prx6 peroxidase activity, the Cys47 in the active center is oxidized to sulfenic acid; this deprives it of activity; as for the reduction of which, a second thiol is required to form a mixed disulfide, then a sulfhydryl group. However, the availability of the sulfenic group is low, due to the peculiarities of the globular structure of Prx6. Prx6 activation occurs during the formation of a heterodimer with GSTP1-1, which promotes the S-glutathionylation of Cys47 Prx6. The conformational changes of the heterodimer occur, providing the formation of a disulfide bond between Cys47 GSTP1-1 and Cys47 Prx6, followed by the reduction of disulfide with the participation of GSH and the regeneration of Cys47 Prx6 [34].
The enzymatic S-glutathionylation of AMP-activated protein kinase (AMPK) occurs not only with the participation of GSTP1-1, but also GSTM1-1 in the absence of strong oxidants (i.e., under conditions which are similar to physiological oxidation). S-glutathionylation occurs at the Cys299 and Cys304 residues and causes conformational changes that activate the kinase activity of human AMPK[32].
The enzymatic S-glutathionylation of AMP-activated protein kinase (AMPK) occurs not only with the participation of GSTP1-1, but also GSTM1-1 in the absence of strong oxidants (i.e., under conditions which are similar to physiological oxidation). S-glutathionylation occurs at the Cys299 and Cys304 residues and causes conformational changes that activate the kinase activity of human AMPK [32].
The ability of S-glutathionylation was found in the enzyme glyoxylase 2 (Glo2). Glo2 hydrolyzes S-
d
-lactoylglutathione to glutathione and lactic acid, while GS
− is formed in the active center of Glo2, similar to GSTP1-1[41]. It has been established that actin and malate dehydrogenase can serve as substrates for S-glutathionylation by Glo2[42].
is formed in the active center of Glo2, similar to GSTP1-1 [41]. It has been established that actin and malate dehydrogenase can serve as substrates for S-glutathionylation by Glo2 [42].
S-glutathionylation is a reversible post-translational modification and, as a rule, deglutathionylation proceeds with the participation of enzymes and is more carefully regulated, in comparison with S-glutathionylation. Glutaredoxin (Grx) is one of the most effective and well-studied enzymes, which reduces Pr-SSG. In the traditional classification, they are divided into mono- and dithiol Grx, depending on whether one or two cysteine residues, respectively, are in the active center. The role of dithiol Grxs is mainly considered in the regulation of reversible S-glutathionylation [26][43][44].
S-glutathionylation is a reversible post-translational modification and, as a rule, deglutathionylation proceeds with the participation of enzymes and is more carefully regulated, in comparison with S-glutathionylation. Glutaredoxin (Grx) is one of the most effective and well-studied enzymes, which reduces Pr-SSG. In the traditional classification, they are divided into mono- and dithiol Grx, depending on whether one or two cysteine residues, respectively, are in the active center. The role of dithiol Grxs is mainly considered in the regulation of reversible S-glutathionylation [26,43,44].
Mammalian dithiol Grxs, Grx1 and Grx2, are found in many cellular compartments; however, Grx1 is mainly present in the cytosol (~1 μM) and mitochondrial intermembrane space (~0.1 μM), while Grx2 is localized mainly in the mitochondrial matrix (~1 μM) [45][46]. Being thiol oxidoreductases, Grx1 and Grx2 contain the CXXC motif (Cys
Mammalian dithiol Grxs, Grx1 and Grx2, are found in many cellular compartments; however, Grx1 is mainly present in the cytosol (~1 μM) and mitochondrial intermembrane space (~0.1 μM), while Grx2 is localized mainly in the mitochondrial matrix (~1 μM) [45,46]. Being thiol oxidoreductases, Grx1 and Grx2 contain the CXXC motif (Cys N
-XX-Cys
C
; CPYC in Grx1 and CSYC in Grx2) in the active site. In addition, they are characterized by the presence of a thioredoxin fold, consisting of four β-sheets surrounded by three α-helices, and a site responsible for stabilizing GSH. Grx uses GSH as a co-substrate for the reduction of Pr-SSG mixed disulfides.
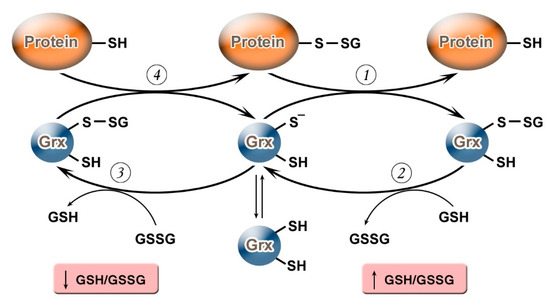
It should be noted that, depending on the value of the GSH/GSSG ratio, Grx can not only carry out deglutathionylation but, on the contrary, may promote S-glutathionylation (
). Grx2 functions as a glutathionylation enzyme under a decrease of GSH/GSSG and an increase in the level of Н
2
О
2
(e.g., in relation to the respiratory complex I) whereas, at a high level of GSH/GSSG, and low concentrations of Н
2
О
2
, Grx2 has a deglutathionylating activity [47]. The putative mechanism of S-glutathionylation proceeds in several stages: first, there is a nucleophilic attack of the disulfide bond GSSG by the thiolate anion Grx-S −
, along with the formation of the glutathionylated intermediate Grx-SSG, from which the activated cationic radical [GS
]
+
is transferred to the target protein with the formation of Pr-SSG, while Grx is again capable of catalyzing the reaction. For this process, the possibility of the reversible formation of Grx-S
2 from Grx-SSG is also noted[48].
from Grx-SSG is also noted [48].
Figure 1.
Glutaredoxin catalytic mechanism in dependence of GSH/GSSH ratio. Under an increase in GSH/GSSG, Grx can catalyze the deglutathionylation of proteins. The glutathionylated sulfur moiety of the protein–SSG is attacked by the thiolate anion of the enzyme (Grx-S
−
), forming the covalent enzyme intermediate (GRx–SSG) and releasing the reduced protein–SH as the first product (1). The second rate-determining step involves the reduction of Grx–SSG by GSH to produce glutathione disulfide (GSSG) as the second product, recycling the reduced enzyme (Grx–S
−
) (2). Under conditions of decreased GSH/GSSG ratio, Grx can catalyze S-glutathionylation of proteins. The S-glutathionylated Grx (Grx–SSG), formed in reaction with GSSG (3), reacts with a protein to create S-glutathionylated protein (protein–SSG) (4).
In addition to Grx, the ability to catalyze deglutathionylation has been observed in some other enzymes (
).
Table 1. Enzymes with capacity to catalyze S-glutathionylation and deglutathionylation.
The isoform of glutathione transferase, GSTO1-1, has the ability to catalyze protein deglutathionylation[54][55]. The isozyme is structurally similar to Grx, including Trx-like folding and a glutathione binding site, where it can form a disulfide bond with a conserved cysteine residue in the active site[53]. Other GST isoforms, including GSTA, GSTM, GSTP, GSTT, GSTS, and GSTZ, in contrast, have catalytic tyrosine or serine residues. In addition, GSTO1-1 has a relatively accessible pocket in the active site, which can potentially accommodate a protein or peptide as a substrate[54][64]. GSTO1-1 catalyzes Grx-like protein deglutathionylation in two similar stages: in the first, the Cys32 of the active site in human GSTO1-1 interacts with Pr-SSG, resulting in reduced Pr-SH and mixed disulfide GSTO1-1-Cys
The isoform of glutathione transferase, GSTO1-1, has the ability to catalyze protein deglutathionylation [54,55]. The isozyme is structurally similar to Grx, including Trx-like folding and a glutathione binding site, where it can form a disulfide bond with a conserved cysteine residue in the active site [53]. Other GST isoforms, including GSTA, GSTM, GSTP, GSTT, GSTS, and GSTZ, in contrast, have catalytic tyrosine or serine residues. In addition, GSTO1-1 has a relatively accessible pocket in the active site, which can potentially accommodate a protein or peptide as a substrate [54,64]. GSTO1-1 catalyzes Grx-like protein deglutathionylation in two similar stages: in the first, the Cys32 of the active site in human GSTO1-1 interacts with Pr-SSG, resulting in reduced Pr-SH and mixed disulfide GSTO1-1-Cys 32S-SG, which is deglutathionylated with the participation of GSH to form GSSG and functional active GSTO1-1, which is capable of catalyzing the deglutathionylation of the next protein substrate[54]. The question of the role that GSTO1-1 plays in the S-glutathionylation of proteins remains open [53].
S-SG, which is deglutathionylated with the participation of GSH to form GSSG and functional active GSTO1-1, which is capable of catalyzing the deglutathionylation of the next protein substrate [54]. The question of the role that GSTO1-1 plays in the S-glutathionylation of proteins remains open [53,55].
The process of deglutionylation is also carried out with the participation of the main members of the Trx family, which play essential roles in maintaining cellular redox homeostasis. Thioredoxins (Trx) 1 and 2 restore disulfide bonds in proteins. This process involves two cysteine residues of the Trx active site (Cys-X-X-Cys), where the disulfide bond is transferred from the substrate protein to Trx. Then, the oxidized Trx is reduced by the NADPH-dependent Trx reductase (TrxR)[65]. In addition, using the mechanism of dithiol reduction, Trxs are able to carry out deglutathionylation without the participation of GSH, which has been shown for glyceraldehyde-3-phosphate dehydrogenase, Prx3, 20S proteasome, and NOS3[59][60][61][62]; however, the exact mechanism of deglutathionylation has not yet been determined.
The process of deglutionylation is also carried out with the participation of the main members of the Trx family, which play essential roles in maintaining cellular redox homeostasis. Thioredoxins (Trx) 1 and 2 restore disulfide bonds in proteins. This process involves two cysteine residues of the Trx active site (Cys-X-X-Cys), where the disulfide bond is transferred from the substrate protein to Trx. Then, the oxidized Trx is reduced by the NADPH-dependent Trx reductase (TrxR) [65]. In addition, using the mechanism of dithiol reduction, Trxs are able to carry out deglutathionylation without the participation of GSH, which has been shown for glyceraldehyde-3-phosphate dehydrogenase, Prx3, 20S proteasome, and NOS3 [59,60,61,62]; however, the exact mechanism of deglutathionylation has not yet been determined.
The ability to deglutathionate proteins has also been observed in protein disulfide isomerases (PDIs), which are also included in the Trx family[63]. However, the significance of the PDI contribution to this process is not yet clear, as their main function is the exchange of the disulfide bonds of PDIs and target proteins. PDIs are enzymes of the endoplasmic reticulum, which are specifically responsible for protein folding through the oxidation of newly formed proteins and isomerization of proteins with improperly formed disulfide bonds, achieving the formation of their native structure. Moreover, PDIs can be secreted by the cell or associated with the cell surface to maintain proteins in a reduced state[62].
The ability to deglutathionate proteins has also been observed in protein disulfide isomerases (PDIs), which are also included in the Trx family [63]. However, the significance of the PDI contribution to this process is not yet clear, as their main function is the exchange of the disulfide bonds of PDIs and target proteins. PDIs are enzymes of the endoplasmic reticulum, which are specifically responsible for protein folding through the oxidation of newly formed proteins and isomerization of proteins with improperly formed disulfide bonds, achieving the formation of their native structure. Moreover, PDIs can be secreted by the cell or associated with the cell surface to maintain proteins in a reduced state [62].
Sulfiredoxin (Srx), for which the ability to reduce the cysteine residue oxidized to sulfinic acid in the active site of typical 2-Cys perxiredoxins (2-Cys Prx) was originally established, is also capable of deglutathionylation, in relation to at least the Prx isoforms[13][56][57], actin, and tyrosine protein phosphatase 1B[58]. For example, it has been shown in vitro that human Prx1 can be S-glutathionylated at three out of four cysteine residues—Cys52, Cys173, and Cys83—and deglutathionylation at Cys83 and Cys173 is catalyzed by Srx, while deglutathionylation at Cys52 is carried out by Grx[56]. The mechanism of Srx-catalyzed deglutathionylation has not yet been fully determined. The data indicate that it proceeds by a mechanism similar to that catalyzed by Grx through the formation of the Srx-SSG intermediate glutathionylated at the conservative Cys99 residue[56]. Srx-catalyzed deglutathionylation appears to have broad substrate specificity. In HEK293 cells transfected with Srx, a decrease in the total content of S-glutathionylated proteins formed under conditions of nitrosative stress after treatment with the nitric oxide donor PABA/NO has been demonstrated[58].
Sulfiredoxin (Srx), for which the ability to reduce the cysteine residue oxidized to sulfinic acid in the active site of typical 2-Cys perxiredoxins (2-Cys Prx) was originally established, is also capable of deglutathionylation, in relation to at least the Prx isoforms [13,56,57], actin, and tyrosine protein phosphatase 1B [58]. For example, it has been shown in vitro that human Prx1 can be S-glutathionylated at three out of four cysteine residues—Cys52, Cys173, and Cys83—and deglutathionylation at Cys83 and Cys173 is catalyzed by Srx, while deglutathionylation at Cys52 is carried out by Grx [56]. The mechanism of Srx-catalyzed deglutathionylation has not yet been fully determined. The data indicate that it proceeds by a mechanism similar to that catalyzed by Grx through the formation of the Srx-SSG intermediate glutathionylated at the conservative Cys99 residue [56]. Srx-catalyzed deglutathionylation appears to have broad substrate specificity. In HEK293 cells transfected with Srx, a decrease in the total content of S-glutathionylated proteins formed under conditions of nitrosative stress after treatment with the nitric oxide donor PABA/NO has been demonstrated [58].
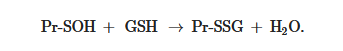 Pr-SOH + GSH → Pr-SSG + H2
Pr-SOH + GSH → Pr-SSG + H2