Microglia are brain-dwelling macrophages and major parts of the neuroimmune system that broadly contribute to brain development, homeostasis, ageing and injury repair in the central nervous system (CNS).
- microglia
- neurodegeneration
- neuroinflammation
- macrophages
- homeostasis
1. Introduction
Microglia are brain-dwelling parenchymal macrophages[1] [1] that are distinct from other brain-dwelling non-parenchymal macrophagic populations and tissue macrophages [2,3][2][3]. Under healthy and normal conditions, microglia are self-maintained with no considerable contribution of peripheral myeloid cells [4][4]. In addition, ~5% of the total cells of the neocortex are normally microglial [5][5]. These macrophages have a uniform distribution throughout the central nervous system (CNS) parenchyma, with a population of approximately 10% of the total cells in the CNS. In addition, they can form a cellular grid with their ramified and highly motile process [6][6]. During the developmental phase of the brain, microglia help shape neural circuits by modulating the strength of synaptic transmissions and sculpting neuronal synapses. When sensing any CNS injury, they become phagocytic and eliminate microbes, cell debris, protein aggregates or any form of CNS insult. However, microglial activation has been reported during several neurological conditions [7,8][7][8], in which they have evidently been playing both beneficial and detrimental roles, depending on disease progression.
The correct functioning of microglia is necessary for neural circuit remodelling and synaptic function, but their involvement in the pathogenesis of neurological diseases raises the debate of whether microglial activation is beneficial or detrimental. In response to CNS insults, microglia proliferate rapidly [9], but their proliferation and turnover rates under homeostatic conditions have not been well documented. Filling this research gap could differentiate them from disease-causing agents [10][10]. Several studies have shown interesting but conflicting results, either very low or very high microglial turnover in mice and the post-mortem brain [11,12[11][12][13],13], although the validity of these techniques has been arguable and limited [14,15][14][15]. However, it is clear that understanding the regulation of microglial populations in the brain is crucial to understanding their functioning at different phases of age and age-related diseases. In one case, the mouse model of AD showed that early priming of individual microglia can induce long-lasting functional changes in life, whereas microglial senescence may have a role in age-related neurodegeneration [10][10]. For example, stimulation of the brain’s immune system during development can induce long-lasting changes in microglial immune responses [16,17][16][17], and ageing and senescence of microglia contribute to neurodegenerative disorders [18][18].
2. Molecular and Functional Background of Microglia
Microglia are the resident immune cells of the brain and cover almost 5–12% of CNS cells [19][19]. In addition, microglia are involved in the homeostasis of host defence against pathogens and consequent neurological disorders [20,21][20][21]. These cells are mesenchymal, originating in the yolk sac, and do not require hematopoietic stem cells for renewal [13,22][13][22]. Their survival and maintenance depend on cytokines, including CSF1 and interleukin (IL)-34 [23][23], and on transcription factors such as IRF8 [22][22]. However, microglia can be simply defined as innate immune cells of the CNS that originate from myeloid cells and express several genes, including Cx3cr1, CD11b, Iba1 and F4/80 [21][21]. Depending on comprehensive knowledge of microglial gene expression and relevant functions [21[21][24],24], this study attempted to determine microglial functions in accordance with their gene expressions. There are three basic functions of microglia—sensing their environment, maintaining physiological homeostasis and protecting against self-modified and exogenous injurious agents. Furthermore, these normal functions are important regulators from a human being’s embryonic stages through to old age.
Sensing is the primary requirement for microglia to function in housekeeping and defending their host from injury (Figure 1). Microglia form a network that spans throughout the CNS. Their thin processes are dynamic and in constant motion, allowing them to scan the area surrounding their cell body every few hours and rapidly polarise toward focal injury [8,19][8][19]. This network includes approximately 100 or more genes that consistently scan surrounding cell bodies and sense any changes in their microenvironment [24,25,26][24][25][26]. The sensome mRNA is expressed uniformly in microglia of different regions of the brain, indicating that all microglia are capable of sensing functions.
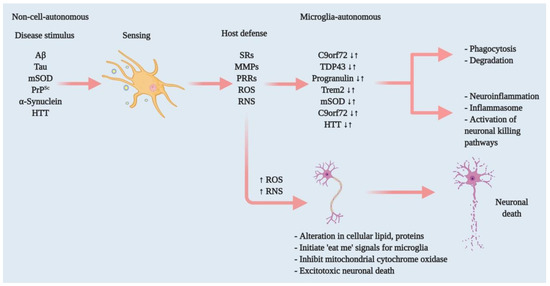
Figure 1. Effectors of microglia function associated with neurodegeneration and how microglia damage or kills neurons. Microglia have both host-defensive and detrimental actions, depending on the scenario. When they encounter aberrant or misfolded proteins—such as Aβ, aggregated α-synuclein, oxidised or mSOD1, or PrPSc—they function as sentinels, protective hosts. In response to these toxic stimuli, microglia attempt to clear aggregated Aβ, α-synuclein or mSOD1 via SRs and other PRRs. However, the nature of the aberrant proteins or their continued production disrupts microglial housekeeping functions. Several pathways activate: NADPH oxidase produces superoxide and derivative oxidants; iNOS produces nitric oxide derivatives, glutamate, cathepsin B and other proteases, and that produces stressed neurons. These processes dysregulate the microglial host-defensive mechanism, leading to an exaggerated proinflammatory response, neurotoxicity and neurodegeneration. Microglia also include tumour necrosis factor (TNF) as an indirect pathway to damage or kill neurons via reducing brain-derived neurotrophic factor (BDNF) and insulin-like growth factor (IGF) production. Neurodegenerative diseases such as AD, ALS and HD, furthermore, cause mutations in specific genes that lead to self-autonomous dysregulation of host defence. This initiates or exaggerates proinflammatory responses, resulting in neurotoxicity and neurodegeneration. In this way, when mutations in TDP-43, progranulin and Trem2 increase (↑), they affect phagocytosis and associated degradation pathways. Similarly, mutations in mSOD and HTT (↑) also affect inflammasome activation and neuronal killing pathways. On the other hand, mutations in C9orf72 can affect both phagocytosis and inflammasome pathways. In normal physiology, a microglia-autonomous mechanism controls the scenario by clearing (↓) mutant or aberrant proteins (adapted from ref. [8][8]).
The second and most important part of microglial function is physiological homeostasis. This function includes synaptic remodelling such as CNS development and maintenance, neurodegeneration [27[27][28],28], phagocytosis of dead or malfunctioning neuronal cells or cell debris [29[29][30],30], or myelin homeostasis [31] [31] (Figure 2). In addition, microglia activate several inflammatory pathways that cause neuroinflammation and possibly neurodegeneration. In this process, microglia interact with astrocytes, and their interaction is also important in regulating homeostasis. Several chemokine and chemoattractant housekeeping genes are involved in phagocytosis (Trem2), synaptic pruning and remodelling (C1q and Cx3cr1) [24], and anomalies in these housekeeping genes may lead to neurodegeneration.
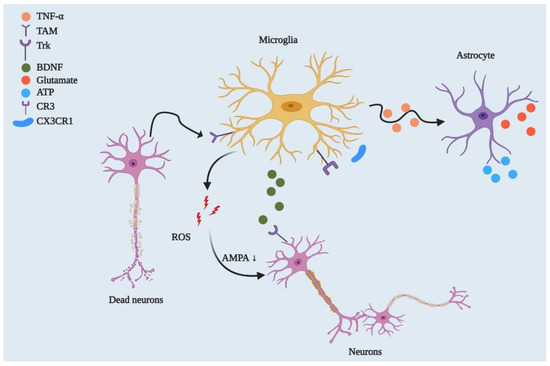
Figure 2. Microglial regulation of neuronal networks and CNS homeostasis. By playing a phagocytic role, microglia engulf dead or degenerated neurons via TAM receptor-mediated recognition of GAS6-opsonised cells. Microglia control synaptic plasticity by downregulating AMPA receptor activity through ROS secretion and by modulating BDNF/Trk receptor pathways. Beyond this, microglia can strip synapses through Cx3cr1-Cx3cl1 interactions and microglial major histocompatibility complex II (MHCII) [32][32]. They can also affect neuronal circuitry via interacting with astrocytes through the TNF-mediated pathway, which releases glutamate and ATP from astrocytes.
As carriers of innate immunity, microglia defend their host against pathogens (Figure 1), injurious proteins including Aβ, aggregated α-synuclein, mutant huntingtin (mHtt), mutant prion (mPrPSc) and oxidised superoxide dismutase (SOD). Microglia activate several receptors to incite host defences, such as expressing Fc receptors, Toll-like receptors (TLRs), and several antimicrobial peptides including Camp and Ngp [24][24]. Therefore, microglia approach the neuroinflammatory threshold by producing peripheral inflammatory cytokines such as TNF-α and IL-1β [33,34][33][34]. In addition, this process involves chemokines (e.g., Ccl2) that recruit additional cells and work together to clear pathogens and normalise the brain in a homeostatic condition [35][35]. However, consistent microglia-induced neuroinflammation leads to neurotoxicity and, eventually, neurodegeneration. In contrast to the anomalies in this microglial functioning, healthy neural microglia are always actively functioning through sensing, housekeeping and protecting their host.
3. Microglia as a Therapeutic Possibility in Neurodegenerative Diseases
Microglia play vital roles at different brain development phases. With age or due to aberrant endogenous or exogenous stimuli, they begin losing their normal physiological functions. Thus, homeostasis in CNS microglia is necessary to disrupt neurodegenerative disease pathology and progression. In addition, identification of critical microglial markers is important to find new therapeutic strategies. Initial studies have suggested an M1 and M2 activation paradigm, in which M1 activation promotes inflammatory cytokines, and M2 activation promotes neurotrophic factor release. However, advanced studies have found that this paradigm does not fit during neurodegeneration. That is, M1/M2 activation does not always function as expected. For instance, in regular cases, M1 activation produces neurotoxicity via proinflammatory release, whereas in some cases, this activation promotes axonal regeneration [118][36]. An AD model study showed that M1 activation promotes Aβ plaque clearance, and, in contrast, M2 activation may ease amyloid spread [119][37].
Microglia are emerging as a cell type used to understand neurodegenerative diseases, but the major challenge is studying human microglia in vitro. For an in vitro neurological disease model study, microglial cells could possibly be developed by differentiating iPSCs or monocytes [120[38][39],121], which has recently been demonstrated. In addition, authors have shown that human microglial-like cells (iMGLs) have phenotypic similarity to in vivo microglia, such as through inflammatory cytokine release or CNS substrate phagocytosis. On the other hand, human monocyte-derived, microglia-like (MDMi) cells have not only presented with microglia phenotype and functions but have also presented with altered expression of gene loci related to neurodegenerative diseases such as AD, PD and MS. These two in vitro microglial cell models could beneficiate therapeutics screening in vitro for neurodegenerative diseases. In addition, genetic defects in microglia could be edited by replacing allogenic or autologous stem cells or monocytes through bone marrow transplantation. Although the latter has not been successfully established for all neurological diseases—though it has for X-linked adrenoleukodystrophy—a recent study showed that brain-engrafted bone marrow derived microglia after a long time in AD mice [122,123][40][41]. It has been suggested that peripheral myeloid cells constitute a heterogeneous cell population that is more effective at clearing Aβ plaque than CNS resident microglia. Extrapolating this therapy with additional triggering could bring success or could be useful for studying other neurological diseases.
Microglial phagocytosis could be another option, but therapeutic agents that target microglial phagocytosis can have both beneficial and detrimental effects—another double-edged sword in neurodegeneration. Microglial phagocytosis opsonises misfolded protein plaques, including Aβ, via the Fc receptor to help antibodies that target misfolded proteins [7,124,125][7][42][43]. In the same way, defective microglia activation in Trem2-deficient mice showed a lack of effectiveness toward the anti-Aβ antibodies [126][44]. Several antibodies currently used in autoimmune diseases may be beneficial in neurodegenerative diseases as well because they target specific proinflammatory cytokines, such as IL-6 and IL-1, or their receptors. Compounds targeting CSF1R can affect proinflammatory microglia activation in AD [127,128][45][46] and reduce microglia-induced inflammation and/or neuronal death[47] [129] in neurodegenerative diseases. In the same vein, IL-34 and CSF1, ligands of CSF1R, may provide neuroprotection and promote neuronal cell survival shown in neurodegenerative models by activating CSF1R in neuron populations but not in microglia [130][48].
In addition, bexarotene-induced Trem2 expression in microglia is, at least in part, mediated by ApoE/Trem2 signalling activation [131][49]. Thus, developing anti-ApoE antibodies in carriers of the ApoE4 allele may help to prevent amyloid deposition and its consequences [132][50]. Moreover, neuronal autophagy has been shown to be useful in neurodegenerative diseases for clearing misfolded proteins and reducing inflammatory cytokines [133][51]. However, very little is known about the role of microglia in autophagy. In one study, loss-of-function mutations of TBK1 affected autophagy in myeloid cells and increased susceptibility to ALS [134][52]. Beyond this, microglial autophagy facilitates Aβ clearance and reduces NLRP3 inflammasome activation [135][53]. Therefore, further study of autophagy in microglia may enhance the understanding of whether drugs activating autophagy have beneficial or detrimental impacts on neurodegenerative diseases.
Reference (Editors will rearrange the references after the entry is submitted)
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393.
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804.
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762.
- Hefendehl, J.K.; Neher, J.J.; Sühs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369.
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2481–2493.
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376.
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415.
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405.
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; Staszewski, O.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803.
- Nowakowski, R.S.; Hayes, N.L. New neurons: Extraordinary evidence or extraordinary conclusion? Science 2000, 288, 771.
- Rakic, P. Neurogenesis in adult primates. Prog. Brain Res. 2002, 138, 3–14.
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660.
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224.
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142.
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170.
- El Khoury, J. Neurodegeneration and the neuroimmune system. Nat. Med. 2010, 16, 1369–1370.
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560.
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280.
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760.
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905.
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519.
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244.
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543.
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935.
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413.
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016, 196, 3375–3384.
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360.
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438.
- Shinjo, R.; Imagama, S.; Ito, Z.; Ando, K.; Nishida, Y.; Ishiguro, N.; Kadomatsu, K. Keratan sulfate expression is associated with activation of a subpopulation of microglia/macrophages in Wallerian degeneration. Neurosci. Lett. 2014, 579, 80–85.
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173.
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.
- Ryan, K.J.; White, C.C.; Patel, K.; Xu, J.; Olah, M.; Replogle, J.M.; Frangieh, M.; Cimpean, M.; Winn, P.; McHenry, A.; et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017, 9.
- Prokop, S.; Miller, K.R.; Drost, N.; Handrick, S.; Mathur, V.; Luo, J.; Wegner, A.; Wyss-Coray, T.; Heppner, F.L. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 2015, 212, 1811–1818.
- Varvel, N.H.; Grathwohl, S.A.; Degenhardt, K.; Resch, C.; Bosch, A.; Jucker, M.; Neher, J.J. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 2015, 212, 1803–1809.
- Wisniewski, T.; Goñi, F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015, 85, 1162–1176.
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56.
- Xiang, X.; Werner, G.; Bohrmann, B.; Liesz, A.; Mazaheri, F.; Capell, A.; Feederle, R.; Knuesel, I.; Kleinberger, G.; Haass, C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. Embo Mol. Med. 2016, 8, 992–1004.
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. BrainBehav. Immun. 2017, 61, 1–11.
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397.
- Olmos-Alonso, A.; Schetters, S.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain A J. Neurol. 2016, 139, 891–907.
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172.
- Savage, J.C.; Jay, T.; Goduni, E.; Quigley, C.; Mariani, M.M.; Malm, T.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6532–6543.
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929.
- Nixon, R.A.; Yang, D.S. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 2012, 4.
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527.
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775.
References
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515.
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845.
- Sheng, J.; Ruedl, C.; Karjalainen, K. Most Tissue-Resident Macrophages Except Microglia Are Derived from Fetal Hematopoietic Stem Cells. Immunity 2015, 43, 382–393.
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804.
- Pelvig, D.P.; Pakkenberg, H.; Stark, A.K.; Pakkenberg, B. Neocortical glial cell numbers in human brains. Neurobiol. Aging 2008, 29, 1754–1762.
- Hefendehl, J.K.; Neher, J.J.; Sühs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369.
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 2481–2493.
- Füger, P.; Hefendehl, J.K.; Veeraraghavalu, K.; Wendeln, A.C.; Schlosser, C.; Obermüller, U.; Wegenast-Braun, B.M.; Neher, J.J.; Martus, P.; Kohsaka, S.; et al. Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat. Neurosci. 2017, 20, 1371–1376.
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience 1992, 48, 405–415.
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405.
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; Staszewski, O.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803.
- Nowakowski, R.S.; Hayes, N.L. New neurons: Extraordinary evidence or extraordinary conclusion? Science 2000, 288, 771.
- Rakic, P. Neurogenesis in adult primates. Prog. Brain Res. 2002, 138, 3–14.
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660.
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224.
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142.
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170.
- El Khoury, J. Neurodegeneration and the neuroimmune system. Nat. Med. 2010, 16, 1369–1370.
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560.
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280.
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760.
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905.
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519.
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244.
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543.
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935.
- Fuhrmann, M.; Bittner, T.; Jung, C.K.; Burgold, S.; Page, R.M.; Mitteregger, G.; Haass, C.; LaFerla, F.M.; Kretzschmar, H.; Herms, J. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2010, 13, 411–413.
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.
- Healy, L.M.; Perron, G.; Won, S.Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016, 196, 3375–3384.
- Pasciuto, E.; Burton, O.T.; Roca, C.P.; Lagou, V.; Rajan, W.D.; Theys, T.; Mancuso, R.; Tito, R.Y.; Kouser, L.; Callaerts-Vegh, Z.; et al. Microglia Require CD4 T Cells to Complete the Fetal-to-Adult Transition. Cell 2020, 182, 625–640.
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360.
- El Khoury, J.; Toft, M.; Hickman, S.E.; Means, T.K.; Terada, K.; Geula, C.; Luster, A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007, 13, 432–438.
- Shinjo, R.; Imagama, S.; Ito, Z.; Ando, K.; Nishida, Y.; Ishiguro, N.; Kadomatsu, K. Keratan sulfate expression is associated with activation of a subpopulation of microglia/macrophages in Wallerian degeneration. Neurosci. Lett. 2014, 579, 80–85.
- Doty, K.R.; Guillot-Sestier, M.V.; Town, T. The role of the immune system in neurodegenerative disorders: Adaptive or maladaptive? Brain Res. 2015, 1617, 155–173.
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.
- Ryan, K.J.; White, C.C.; Patel, K.; Xu, J.; Olah, M.; Replogle, J.M.; Frangieh, M.; Cimpean, M.; Winn, P.; McHenry, A.; et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017, 9.
- Prokop, S.; Miller, K.R.; Drost, N.; Handrick, S.; Mathur, V.; Luo, J.; Wegner, A.; Wyss-Coray, T.; Heppner, F.L. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer’s disease-like mice. J. Exp. Med. 2015, 212, 1811–1818.
- Varvel, N.H.; Grathwohl, S.A.; Degenhardt, K.; Resch, C.; Bosch, A.; Jucker, M.; Neher, J.J. Replacement of brain-resident myeloid cells does not alter cerebral amyloid-β deposition in mouse models of Alzheimer’s disease. J. Exp. Med. 2015, 212, 1803–1809.
- Wisniewski, T.; Goñi, F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron 2015, 85, 1162–1176.
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56.
- Xiang, X.; Werner, G.; Bohrmann, B.; Liesz, A.; Mazaheri, F.; Capell, A.; Feederle, R.; Knuesel, I.; Kleinberger, G.; Haass, C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. Embo Mol. Med. 2016, 8, 992–1004.
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. BrainBehav. Immun. 2017, 61, 1–11.
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397.
- Olmos-Alonso, A.; Schetters, S.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain A J. Neurol. 2016, 139, 891–907.
- Luo, J.; Elwood, F.; Britschgi, M.; Villeda, S.; Zhang, H.; Ding, Z.; Zhu, L.; Alabsi, H.; Getachew, R.; Narasimhan, R.; et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med. 2013, 210, 157–172.
- Savage, J.C.; Jay, T.; Goduni, E.; Quigley, C.; Mariani, M.M.; Malm, T.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 6532–6543.
- Luz, I.; Liraz, O.; Michaelson, D.M. An Anti-apoE4 Specific Monoclonal Antibody Counteracts the Pathological Effects of apoE4 In Vivo. Curr. Alzheimer Res. 2016, 13, 918–929.
- Nixon, R.A.; Yang, D.S. Autophagy and neuronal cell death in neurological disorders. Cold Spring Harb. Perspect. Biol. 2012, 4.
- Ahmad, L.; Zhang, S.Y.; Casanova, J.L.; Sancho-Shimizu, V. Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med. 2016, 22, 511–527.
- Cho, M.H.; Cho, K.; Kang, H.J.; Jeon, E.Y.; Kim, H.S.; Kwon, H.J.; Kim, H.M.; Kim, D.H.; Yoon, S.Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775.